Through the Looking Glass of Single-Cell Proteomics

Complete the form below to unlock access to ALL audio articles.
Out of the enormous number of proteins in a cell (mammalian cells are estimated to have about 10 billion proteins per cell), being able to study even a handful has helped scientists decipher many biological mysteries. But many more remain to be solved, and with the growing demands of precision medicine, the field of single-cell proteomics has had to mature and adapt quickly.
Every day, researchers are tirelessly working to develop new methods and techniques that allow the comprehensive profiling of hundreds to thousands of proteins in individual cells, at an acceptable throughput. Thanks to these industrious efforts, our understanding of normal physiology, as well as how diseases can develop and progress, has advanced significantly.
What’s more, investigations into the proteome of stem cells have led researchers to identify certain proteins that play a central role in stem cell differentiation. By studying these proteins, it may be possible to control and direct the differentiation of stem cells, which has huge implications for regenerative medicine.1
In this article, we take a closer look at how several scientists in North America are using single-cell proteomics (SCP) technologies to discern disease pathogenesis and enhance directed stem-cell differentiation.
It Takes All Kinds of Cells (and Their Proteins) to Make to a Disease Happen
In Assistant Professor Jia Guo’s lab, housed in the School of Molecular Sciences, Arizona State University, Tempe, AZ, USA, the main focus of research is to develop new genomics and proteomics technologies that can analyze a large number of different biomolecules in single cells of structured tissues. Through their work, Guo and his teammates are amassing valuable information on the complex molecular mechanisms of diseases like cancer and neurodegenerative diseases.“We want to know the disease inside out, or as much as we can about how it develops,” said Guo. “The ability to profile proteins comprehensively in single cells is crucial for this knowledge. And only through this knowledge can we improve diagnosis and personalized therapy.”
The inherent complexity in biological systems comes from the many different types of structurally and functionally different cells that make up the systems.2 Such cellular heterogeneity is even more pronounced in disease states, especially cancer. To pick apart the differences between individual cells in complex multicellular organisms, we need to look at cells one-by-one.
“Unlike bulk cell analysis, single-cell analysis can reveal cell heterogeneity, spatial organization, gene expression regulation, and interactions of diverse cell types,” Guo explained.Guo’s group is applying antibody-based SCP technologies to profile neurons, astrocytes and other cells in the human brain. Their work uncovered that neurons in the human hippocampus can be partitioned into different neuron clusters based on their comprehensive protein expression profiles. Furthermore, they found that different subregions of the hippocampus are composed of neurons from different clusters. Next, Guo is ready to “profile the different cell types in Alzheimer’s brains, and compare the cell type composition and their 3-D organization differences between normal and Alzheimer’s brains, to explore molecular mechanisms of Alzheimer’s disease.”

Guo’s lab uses antibody-based SCP technologies to study the human brain: Three proteins are sequentially stained with cleavable Cy5 labeled antibodies in brain tissues. Nuclei are stained with DAPI (blue). Scale bars, 50 μm.
Proteomics to Direct Stem Cell Differentiation
Because of their ability to self-renew and differentiate into multiple cell lineages, stem cells have long been the subject of rigorous study and much excitement in science and medicine. Being able to direct the differentiation of stem cells can help scientists tap into the vast potential these cells present for regenerative medicine.Current in vitro directed differentiation protocols rely on the valuable, but limited, knowledge we have on signaling pathways involved in the development of specific cells and tissues in vivo. While this knowledge – acquired over decades – has provided very useful guidelines, we have much more to learn, in order to understand the making of every cell type.
“This gap in knowledge stems in part from difficulties in quantifying the signaling pathways in the single cells that make up the highly heterogeneous human tissues,” explained Nikolai Slavov, Assistant Professor at the Department of Bioengineering in Northeastern University, Boston, MA, USA.
Why is it so important to look at individual cells? Because interpreting population-average protein levels is fundamentally confounded when samples consist of heterogeneous cells, clarified Slavov. The most obvious caveat is that the population-average may not be representative for any one cell type.
“For example, proteins may have bimodally distributed abundances within the whole heterogeneous cell population, but the average-population levels will correspond to no cell type,” remarked Slavov. “Furthermore, trends within different cell types, may disappear or even reverse when these groups are combined, as with population-average measurements.”Recently, single-cell RNA-seq methods have started to characterize the transcriptional heterogeneity of differentiating single cells, but the levels of RNA are still not enough to characterize post-transcriptional regulation and signaling pathways.3 “So the measurement of protein levels and signaling dynamics in single cells is essential to determine and then engineer the signals required to direct cells to differentiate into different cell types.”
Last year, Bogdan Budnik, Director of Proteomics, Mass Spectrometry & Proteomics Resource Lab at Harvard University, worked with Slavov's team to develop a mass spectrometry (MS)-based single-cell proteomics technology called ‘SCoPE-MS’ to quantify over a thousand proteins in differentiating mouse embryonic stem cells.4 The single-cell proteomes enabled Budnik and Slavov to deconstruct cell populations and determine protein abundance relationships. When single-cell proteomes and transcriptomes were compared, they saw that mRNA covariation appeared to be predictive of protein covariation, even in single cells.4
“But interestingly, many genes showed distinct regulatory patterns at the mRNA and the protein levels,” said Slavov. “This suggests that post-transcriptional regulatory mechanisms play a big role in proteome remodeling when lineages are specified, especially for developmental genes.”
Recent Advancements in Throughput and Sensitivity in Proteomics
As incredible as the potential of SCP is, there are still challenges associated with its usage. Guo pointed out that the low abundance of proteins in individual cells and the inability to amplify proteins are major hurdles in antibody-based SCP. Additionally, the low multiplexing capacity of conventional assays, such as immunofluorescence and immunohistochemistry, presents another challenge.2“Recently, we have seen the development of several microchip, mass cytometry and cyclical immunofluorescence-based approaches that can overcome these issues,” remarked Guo. “Some of the new, super-sensitive assays allow dozens of proteins to be quantitatively profiled in single cells.”
But we can’t stop there. According to Guo, even ‘dozens of proteins’ are not enough. They only represent a small fraction of the whole proteome. Thus, the multiplexing capacity of the current technologies needs to be expanded. In addition, to quantitatively profile the low-expression proteins in clinically-archived tissues (such as formalin-fixed paraffin-embedded (FFPE) tissues), the detection sensitivity of the existing assays has to be significantly enhanced. Factoring in the time element, it would be desirable to further improve the sample throughput. Finally, Guo added that new image analysis algorithms should be developed for more precise cell segmentation and for studies of cell-cell communication.
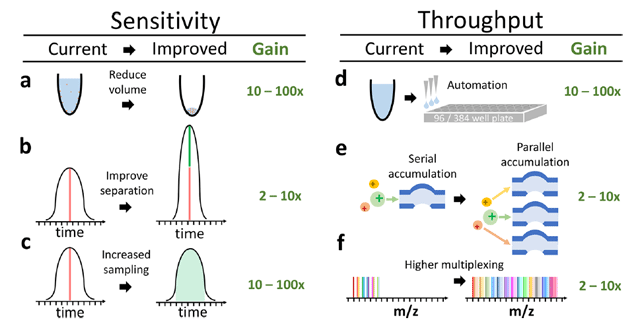
The sensitivity and throughput of single-cell proteomics by mass-spec are poised to grow by orders of magnitude thanks to improvements in sample preparation, automation, peptide separation, multiplexing and instrumentation. (Source: Specht & Slavov, JPR 2018)
Agreeing with Guo, Slavov noted that his team is working on optimizing single-cell MS methods through efficient sample preparation in pure water5, experimental designs that allow multiplexing and enhanced peptide sequencing,4 and better computational strategies for the analysis of mass-spec data.
In conclusion, he added that SCP can essentially expedite the development of regenerative therapies. “Many current approaches to developing directed differentiation rely on trial-and-error screens. This is where SCP becomes really useful. SCP technologies can systematically identify and prioritize screening efforts, and hence speed up the development process.” As Guo and Slavov both indicated in their final remarks, SCP no doubt holds great promise to unlock major mysteries in neuroscience, cancer, and stem cell biology.
UPDATE: This article has been corrected to reflect the involvement of Dr. Bogdan Budnik's team at Harvard University in the development of the SCoPE-MS method. The paper which describes the method has been published in Genome Biology on Oct. 22, 2018.
References:
1Muñoz, J.; Heck, A. J., Perspectives in stem cell proteomics. Genome Medicine 2009, 1 (4), 45.
2Mondal, M.; Liao, R.; Guo, J., Highly Multiplexed Single‐Cell Protein Analysis. Chemistry – A European Journal 2018, 24 (28), 7083-7091.
3Franks, A.; Airoldi, E.; Slavov, N. Post-transcriptional regulation across human tissues. PLoS Computational Biology 2017, 13(5).
4Budnik, B.; Levy, E.; Harmange, G.; Slavov, N., Mass-spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. bioRxiv 2018 (preprint before peer-review).
5Specht, H.; Harmange, G.; Perlman, D. H.; Emmott, E.; Niziolek, Z.; Budnik, B.; Slavov, N., Automated sample preparation for high-throughput single-cell proteomics. bioRxiv 2018.

