Advances in Immunomodulators for Vaccines To Prevent and Treat Autoimmune Diseases

Complete the form below to unlock access to ALL audio articles.
Autoimmune conditions: A worldwide health crisis
Epidemiological studies suggest that environmental rather than genetic factors are responsible for the current autoimmune epidemic. One leading factor is the eradication of parasitic helminths during the past 40 years as a consequence of improved public health care. This change correlates with the rise in autoimmune diseases and highlights the survival strategy developed by parasitic helminths during their evolution with their hosts. The autoimmunity epidemic, initially limited to the industrialized countries, is now spreading worldwide as public health improves in the developing nations. Due to the injurious effects and economic burdens of these diseases, this situation could disrupt the social fabric of these countries.
Mammalian mechanisms are designed to prevent fetal rejection and as a side effect many autoimmune conditions improve during pregnancy, but relapse after birth. This strategy is also used by cancer cells to prevent anti-tumor pro-inflammatory responses. To survive in a mammalian host, helminths’ mimic these mechanisms, biasing the host’s immune system to an anti-inflammatory Th2 immune response to evade destruction while inducing tolerance in the host. Indeed, Th2 immunity characterized by production of anti-inflammatory cytokines and antibodies, apparently was developed to repair cellular damage caused by the pro-inflammatory Th1 immune response. Consequently, helminthiasis has been proposed as a treatment for autoimmunity, but due to the public health hazards it poses it is an unfeasible proposition. Among other suggested methods1 are those based on clonal deletion, vaccines, nanoparticles and cell-based therapies with genetically modified cells. But genetic therapies have many unresolved risks, and regardless of their success, their high cost would limit their use to affluent countries. Currently there are an estimated 50 million autoimmunity cases in the United States, 70 % of which are in women, a number that is on the increase and which highlights the magnitude of the problem. Therefore, a practical approach to treat autoimmunity would be active immunotherapy or vaccination, to reestablish tolerance to those self-antigen(s) liable to induce disease.
Prevention and treatment of autoimmune conditions
Autoimmunity may be triggered by a pathogen if it exposes cryptic self-antigens or has antigens that cross-react with self-antigens. This situation generates a damaging T-cell mediated immunity against a self-antigen(s), a pro-inflammatory Th1/Th17 immune response worsened by “epitope spreading,” where the response extends to other antigens, aggravating autoimmunity2. Therefore, therapies and prevention of autoimmune diseases need to induce Th2 immunity to replace the pro-inflammatory reactions and avert tissue and organ damage. Scientists have looked at vaccination as one approach to address the problem, but efforts have so far been unsuccessful. The reason for these poor results may be the vaccine formulations used. Although much attention has been given to the vaccines’ antigen component, little has been given to the vaccine adjuvants, i.e. the ingredient responsible for inducing a tolerant or non-tolerant immune response against the antigen3. Nonetheless, inhibition of pro and anti-inflammatory immunities can be attained by i) global immunosuppression using inhibitors of DNA or protein synthesis, or ii) check-point based immunotherapy, that blocks specific immune pathways with biologics, decreasing but not eliminating immunosuppression4. Unfortunately, these methods increase vulnerability to cancer and infections.
An alternative to immunosuppressive therapy would be to induce an antigen-specific anti-inflammatory immunity; a strategy used by helminths to establish tolerance. This approach would require sub-unit vaccines which have a self-antigen(s), plus anti-inflammatory adjuvants that induce Th2 while inhibiting Th1 and Th17 immunities, i.e. a sole anti-inflammatory immunity. While many self-antigens that stimulate autoimmunity are available, the lack of anti-inflammatory adjuvants have been an obstacle to vaccine development3. This situation is worsened by the fact that alum5 and oil/water emulsions3, the regularly used adjuvants in these vaccines, while presumed to be purely anti-inflammatory, are not. Actually, depending on the disease, delivery mode and other factors, these adjuvants may also elicit a pro-inflammatory immunity. This may explain why vaccines that have these adjuvants fail in the treatment of autoimmunity, but are successful in infectious diseases, the latter typically requiring a pro-inflammatory response to be effective.
Immunotolerance and dendritic cells
Immunotolerance, the inability to mount an immune response against an antigen, cannot be induced by immunosuppression which shuts down the entire immune system6. Consequently, failure of the “natural” immunotolerance for self-antigens, may cause pro-inflammatory autoimmunity. Attempts to restore “natural” immunotolerance by causing clonal anergy and/or deletion of autoreactive T cells, have thus far yielded poor clinical results3. Hence, an alternative would be to build an “induced” immunotolerance against self-antigens. Here, T-cell mediated autoimmunity would be substituted by an anti-inflammatory immunity; a process that starts at the dendritic cell (DC) level. DCs are antigen presenting cells that initiate and control immunotolerance, by modulating the effector T cell (Teff) responses. Phenotypically, DCs can be sorted into immature (iDC), mature (mDC) and tolerogenic (tDC) phenotypes7. Although DCs’ differentiation is complicated, several critical pathways have been partially elucidated. Exposure of iDCs to exogenous antigens and “danger” signals, initiates a differentiation process to become mDCs, capable of stimulating T cells toward pro-inflammatory immunities. Hence, mDCs show upregulated production of MHC, costimulatory molecules and pro-inflammatory cytokines. Under certain conditions, iDCs differentiate into tDCs, which stimulate the production of regulatory T cells (Tregs) that induce tolerance, suppressing T cell immunity and preventing autoimmunity.
Differentiation into tDCs can be attained using i) biologics like cytokines, e.g. IL-10, ii) pharmacological agents, like rapamycin, and iii) genetic modifications of DCs; methods that may entail complex processing. Remarkably, helminths induce host tolerance by switching Th1/Th17 immunities to Th2, using the DC’s lectin receptor DC-SIGN8,9. This receptor, by binding mannose or fucose, induces a maturation to mDC or m*DCs, respectively; where m*DCs are Th2-associated DC subsets9. Consequently, fucosylated glycans play a critical role in inducing tolerance, as with fetal or tumor tolerance, where LewisX antigen and LNFPIII related glycans bind to DC-SIGN. This binding allows, by receptor-mediated endocytosis, the delivery of the antigen for intracellular processing by DCs. Differentiation of iDCs to m*DCs requires the activation of TLR4 by lipid A and the concurrent signaling of DC-SIGN, all in the presence of IL-410,11. Thus, m*DCs would upregulate expression of anti-inflammatory cytokines, like IL-10, while downregulating pro-inflammatory cytokines, like IL-2 and IL-17, yielding Th2-inducing DCs. Naïve T cells exposed to Th2-inducing m*DCs plus IL-4 will promote a Th2 cell immunity (Fig. 1).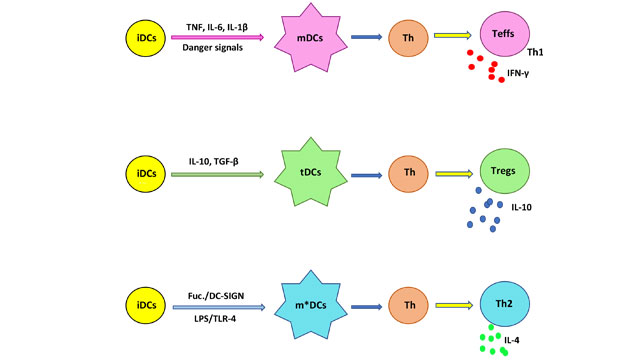
Figure 1. Maturation of iDCs into different subsets. Exposure to danger signals, e.g. TLR ligands, and proinflammatory cytokines, favors the production of mDCs, which would interact with T helper cells (Th) to yield Teffs, that are Th1 proinflammatory cells secreting IFN-γ and other cytokines. iDCs in the presence of IL-10 and TGF-β will mature into tDCs, that polarize Th cells to Tregs, characterized by secretion of the anti-inflammatory cytokine IL-10.The iDCs simultaneously stimulated by lipid A (LPS) of TLR-4 and signaling of DC-SIGN by a fucosylated ligand (Fuc.), would mature into an m*DC which polarizes Th to become Th2 cells with secretion of IL-4, which is another anti-inflammatory cytokine.
DC-SIGN fucosylated ligands, inducers of immunotolerance
The role of fucosylated ligands in inducing immunotolerance is demonstrated by fetal and tumor tolerance; where the fucosylation process is upregulated. Additional proof is provided by animal studies with the myelin oligodendrocyte glycoprotein (MOG), where removal of fucose from this glycoprotein resulted in an increase in the strongly pro-inflammatory Th17 immunity12. Other examples are the suppression of experimental autoimmune encephalomyelitis in a mouse model for multiple sclerosis (MS)13, and the prolonged heart transplant survival in mice, which resulted in Treg cell accumulation14, both upon administration of LNFPIII. Thus, the epidemiological and experimental evidence confirms fucose’s role in promoting immunotolerance. As fucose binding to DC-SIGN polarizes immunity towards Th2 in an antigen-specific manner, autoimmune diseases may be treated and prevented with vaccines that have self-antigens and fucosylated glycans as Th2 adjuvants. It is also important that fucosylated glycans induce immunotolerance but without immunosuppression. The fact that co-delivery of antigens and fucosylated glycans to DCs favors the production of antibodies with enhanced affinity, explains why covalently linking fucosylated glycans to antigens elicits a better Th2 response. Paradoxically, despite DC-SIGN8 being essential for DCs’ differentiation and the main receptor for fucosylated ligands that induce immunotolerance, there are no major drug development efforts focusing on this receptor as an autoimmunity therapeutic target.
To address this need, a new fucosylated adjuvant, QT-0101, has been developed. QT-0101, the deacylated derivative of the pro-inflammatory adjuvant QS-21, is a well-defined glycoside preparation, carrying a fucose residue unrelated to that of LewisX antigen, but capable of biding to DC-SIGN15 (Fig. 2). As QT-0101 elicits an antigen-specific Th2 immunity with inhibition of Th1/Th17 immunities and production of Tregs15,16, it is relevant to vaccines for autoimmune disease. Because of its amphiphilicity, QT-0101 can form complexes with proteins, which allows delivery of these antigens to those DCs being activated and differentiated toward the m*DC phenotype. Its detergent-like properties would facilitate the early escape of the peptides from proteins being processed at the endosomes, favoring a broader array of conformational epitopes. Like fucosylated glycans, QT-0101 induces Tregs16 and, while inhibiting pro-inflammatory immunities, it does not abrogate them. Hence, as with other classic vaccines, maintenance of immune tolerance would require periodic immunizations to boost the immune system. Pausing of vaccination, will halt tolerance and allow the development of the protective pro-inflammatory immunity. The ability to restore pro-inflammatory immunity when required, necessary to fight infection and cancer, could therefore also be a desirable aspect to the strategy, a process that could be reversed by restarting vaccination once the situation had been resolved. Also, QT-0101’s toxicity is much lower than that of its precursor QS-21, an adjuvant approved for human use. Due to cost and convenience, vaccines are valuable options to biologics to treat autoimmune conditions. Yet, they may be also be useful to prevent autoimmune conditions, as in cases where known genetic risk factors exist. QT-0101 is also a suitable adjuvant for vaccines against proteopathies, like Alzheimer’s disease17; which require an anti-inflammatory antibody response, rather than a damaging pro-inflammatory response.
Figure 2. Structures of the fucosylated glycan LNFPIII and glycoside QT-0101. The fucosyl residue (Fuc), shown in blue, has its hydroxyl groups 3 and 4 free for binding to DC-SIGN. The triterpene nucleus of QT-0101 is shown in magenta. The other glycosyl residues are shown in black.
Considering the body of information concerning QT-0101’s structure-activity relationships, the application of medicinal chemistry methods3,17 to this compound would allow the development of new anti-inflammatory agents. Further work in this area is important as the scarcity of these compounds has been the main reason for the lack of progress in active immunotherapy for autoimmune diseases and proteopathies.
References
1. Carballido JM and Santamaria P. 2019. J. Exp. Med. 216:247-250
2. Atassi MZ and Casali P. 2008. Autoimmunity 41:123-132
3. Marciani DJ. 2017. Autoimmunity 50:393-402
4. Li P et al. 2017. Front Pharmacol. 8:460. doi: 10.3389/fphar.2017.00460
5. Kooijman S. et al. J. 2018. Proteomics 175:144-155
6. Bluestone JA. Immunol. Rev. 2011. 241:5-19
7. Maldonado RA and von Andrian UH. 2010. Adv. Immunol. 108:111-165
8. Geijtenbeek TBH and Gringhuis SI. 2009. Nat. Rev. Immunol. 9:465-479
9. Hussaarts L.et al. 2014. Front Immunol. 5:499. doi: 10.3389/fimmu.2014.00499
10. Tundup S. et al. 2012. Ann N.Y. Acad Sci. 1253:E1-E13
11. Gringhuis SI. et al. 2014. Nat Commun. 5:3898
12. Garcia-Vallejo JJ. et al. 2014. J Exp Med. 211:1465-1483
13. Zhu B. et al. 2012. Clin Immunol. 142:351-361
14. Partha D. et al. 2010. Transplantation 90:1071-1078
15. Marciani DJ. 2014. Drug Discov. Today 19:912-920
16. Heidari AR. 2019. et al. Mult Scler Relat Disord. 29:68-82
17. Marciani DJ. 2019. Research. DOI: 10.34133/2019/5341375

