Weighing-up the Pros and Cons: Is There Proteomic Life Without Quantitative LC-MS?

Complete the form below to unlock access to ALL audio articles.
In the very early beginnings of proteomics, there was an overwhelming degree of excitement and enthusiasm as to what mass spectrometry could do for biology and the scientific community as a whole. Initial publications focused primarily on “numbers” of identified proteins and little attention was paid to determining alteration in protein quantities under specified experimental conditions, e.g., perturbation with a drug, or stimulation with a growth factor. One technique that enabled simultaneous separation and quantitation of many proteins, however, was two-dimensional gel electrophoresis (2DE)1. Here, quantitation was achieved by comparative analysis of the protein staining between two or more gels that were representative of cells from different states/treatments. Subsequently, proteins from gel spots of interest were identified by mass spectrometry. The downside of 2DE was that the technique was highly-selective for abundant proteins and the detection of low-abundance regulatory proteins, such as kinases and transcription factors, was limited2.
After the initial excitement over the possibilities of proteomics passed, many biologists and mass spectrometrists alike took a step back and reflected. Would it make more sense to not only correctly identify the proteins in a sample but also to add a quantitative component to each and every protein that was identified? Indeed, the questioning, emergence and eventual acceptance of this paradigm was welcomed by the community; and with the successful coupling of nano-high performance liquid chromatography (nano-HPLC) with nano-electrospray ionization tandem mass spectrometry (nanoESI-MS/MS), came the era of quantitative liquid chromatography mass spectrometry (LC-MS). When asked his opinion on why this area of science is broadly important and critical for researchers, Michael J. McCoss, Professor of Genome Sciences at the University of Washington answers, “It is a field that is used in everything from measuring drugs of abuse, pharmacodynamics, proteomics, metabolomics, environmental pollutants, newborn screening, vitamin D, forensics, etc.” Such a remarkable diversity of applications has ensured that the technological developments and widespread acceptance of quantitative LC-MS has become integral to the daily research of scientists. For proteomic applications in particular, Professor Bernhard Kuster, Chair of Proteomics and Bioanalytics at the Technical University of Munich notes that, “Quantitative measurements are important in proteomics as these aid in objectively analyzing a (biological) system.”
How can I quantitate my protein?
The techniques available for quantitative LC-MS largely fall into two categories: (i) relative quantitation; and (ii) determining peptide abundance by calibrating to a synthetic stable isotope-labeled standard (Figure 1). Currently, the majority of the scientific community apply the former approach to research questions. The latter, however, is beginning to play an ever-increasingly important role; particularly in clinical applications.
Discovery-based relative quantitation by LC-MS has enabled researchers to simultaneously determine changes in protein abundance across multiple samples. As Bernhard Kuster says, “LC-MS is particularly strong in the sense that it can quantitate a lot of proteins in parallel and without having to know a priori which proteins one may want to analyze”. To truly grasp the function of an individual protein and the relationship that protein has with others in the context of a complex biological system, changes in protein abundance relative to those of the baseline or standard system must be measured. As the name indicates, relative quantitation involves the comparison of quantities of proteins against a designated condition, i.e., how much of a peptide (and by extrapolation, a protein) is there in condition B, C, D etc. compared to the quantity of that same peptide in condition A. Kuster also points out that, “LC-MS is also powerful in the sense that quantitative assays can be developed for proteins for which no antibodies exist or are of poor quality”.
In broader terms, there are basically two approaches for relative quantitation of proteins by LC-MS. These are: (i) labeled; and (ii) label-free. The former category is sub-divided into two means of labeling peptides/proteins in a proteome. Namely, via metabolic labeling that was initially pioneered in 19993 and later more widely adopted by the research community in the form of SILAC (stable isotope labeling with amino acids in cell culture)4. Proteins can also be modified by chemical derivatization and such approaches have encompassed isotope-coded affinity tags (ICAT)5, 18O labeling6, dimethyl labeling7, and isobaric mass tags, i.e., tandem mass tag (TMT) and isobaric tag for relative and absolute quantitation (iTRAQ)8,9.
With SILAC, biological samples are labeled in vitro with a heavy isotope version of the target amino acid. During protein synthesis, the naturally-occurring amino acid is replaced by a heavier labeled version. Cells exposed to different experimental conditions (cultured in light, medium, or heavy media) can then be mixed and all processing steps performed on the combined sample (Figure 1, metabolic labeling). This strategy markedly diminishes any variability that can occur during sample preparation and has the distinct advantage of more accurate quantitation. With e.g., TMT reagents8, high-throughput multiplexed protein quantitation can be achieved. When combined with LC-MS and proteomic data software, currently, up to 16 different samples derived from cells, tissues or biological fluids can be simultaneously analyzed, the peptides/proteins identified, and the relative quantities of the peptides/proteins calculated (Figure 1, chemical labeling).
Label-free quantitation by LC-MS has seen a recent resurgence in popularity. This renewed interest from the community has primarily developed as a consequence of improved mass resolution of current mass spectrometers, improved retention time stability with new HPLC systems and improved data analysis algorithms that not only align multiple chromatographic traces; but can also extract numerous features from the traces. In parallel, data-independent acquisition (DIA) has also emerged as a powerful approach for deep, label-free relative protein quantitation across the entire proteome. Overall, a label-free quantitative approach is a very cost-effective alternative to both SILAC and TMT and has the major advantage of comparing alterations in protein abundance across multiple samples without the use of any isotopic labels. Samples are individually analyzed by LC-MS; an aspect that is highly-advantageous in the analysis of cohorts comprised of hundreds to even thousands of patient samples. With sophisticated software algorithms, it is now considerably more straightforward to integrate signals that correspond to unique peptide ions over the entire LC time scale. Post-LC-MS acquisition of the data, the individual analyses can be compared and chromatographic features common to all samples can be aligned (Figure 1, label free). The major advantages of proteome quantitation by label-free LC-MS are the ability to compare an unlimited number of samples, and the obsolescence of expensive labeling strategies. A major disadvantage, however, is that there is increased LC-MS acquisition time (as each sample is run consecutively). Nevertheless, new systems are appearing on the market than can provide very rapid and short LC gradients to diminish the time required to analyze thousands of clinical samples.
The concept of using stable-isotope synthesized peptides as internal standards for quantitation of proteins by LC-MS was first pioneered by Desiderio and Kai10 (Figure 1, spiked standards). These are synthetic tryptic peptides that have the same amino acid sequence as the naturally-occurring peptides of interest. The synthesized peptides, however, contains at least one stable isotope-labeled amino acid that leads to a small increase (6-10 Da) in molecular mass. A ‘known’ quantity of the synthetic peptide equivalent is spiked into a proteomic digest and the entire sample analyzed by LC-MS. The native peptide and the spiked peptide standard will chromatographically co-elute and ionize in the mass spectrometer. The m/z of the two peptides, however, can be easily distinguished. From the extracted ion chromatograms and calculation of area-under-the-curve for both peptides, an estimate of the quantity of the native peptide can be calculated by comparing the peak ratios. The generally-accepted recommendation is to synthesize a minimum of three tryptic peptides per protein. If quantitation of several proteins is required per experiment; however, the price soon escalates. Additionally, although the spiking of peptide standards is quite a straightforward approach, the calibration of peptide abundance to reflect the protein level remains a challenge11. Alternative calibration methods to address these difficulties have been proposed and include, e.g., the use of a single-point common reference pool12,13.
Why should I quantitate my proteins?
Peptide and protein quantitation are central to understanding the dynamics of proteins, especially in the ever-changing flux of the active and dynamic proteome and is fundamental to the research of many key opinion leaders in the proteomic field. Bernhard Kuster routinely uses quantitative LC-MS to further his research. “We aim to better understand how cancer drugs work in cells and tumors. In order to capture the molecular complexity of these biological systems, we need technology that can measure many proteins quantitatively in a single experiment and with high throughput. In this context, all of our research starts with LC-MS on such biological systems in order to build hypotheses that can be followed-up by more specialized experiments.” Similarly, Mike McCoss echoes the words of Kuster and says, “My lab. is an LC-MS lab. Everything we do revolves around making quantitative measurements with LC-MS. We definitely didn't invent it, but we contribute to it and we routinely apply it to lots of different applications.”
When asked about the level of awareness of quantitative LC-MS technology by other scientists, McCoss replies, “I'm pretty sure that all chemists, biochemists and cell biologists, etc. are very familiar with quantitative LC-MS and most have probably benefited or relied on it at some point in time.” Kuster replies in agreement, “Within the life science community, LC-MS as a quantitative approach is very widely-known; however, LC-MS as a whole is still not nearly as often used as genomic technologies. This is because the technology is still not as easily-/widely-accessible to scientists.”
Quantitative LC-MS is fundamental to modern-day proteomics
The technology of quantitative LC-MS is here to stay and has become an integral and important component of any proteomic experiment. Indeed, quantitative measurements are at the core of practically every single proteomic study that is undertaken today. This has been driven by developments in LC-MS and associated sample preparation, separation, and data analysis methods, and LC-MS is now the de facto standard for quantitative measurements in proteomics. For the McCoss group, it is self-evident, “We are predominantly a proteomic lab with a keen interest in developing and applying mass spectrometry for the quantitative analysis of proteins.” The Kuster laboratory runs a reasonably large LC-MS platform comprised of seven systems than operate 24 hours a day, seven days a week. Thus, as Kuster states, “The technology is at the heart of what we do on a daily basis”. Furthermore, “The technology continues to develop rapidly (as does the accompanying informatics). Hence, the perspectives of LC-MS are very bright. What needs to happen is that the technology becomes more accessible to basic life scientists and clinicians in order to make a stronger impact on, e.g., the healthcare sector.”
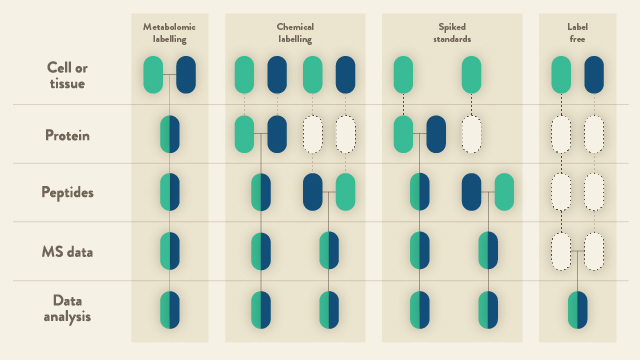
Figure 1: Proteomic workflows based on quantitative LC-MS. Blue and green boxes represent two experimental conditions. Horizontal lines indicate where samples are combined. Dashed lines indicate where experimental variation and, subsequently, quantitative errors can occur. Adapted from14.
References
1. P. H. O'Farrell, J. Biol. Chem. 250, 4007-4021 (1975).
2. S. P. Gygi et al., Mol. Cell. Biol. 19, 1720–1730 (1999).
3. Y. Oda et al., Proc. Natl. Acad. Sci. USA 96, 6591-6596 (1999).
4. S. E. Ong et al., Mol. Cell. Proteomics 1, 376-386 (2002).
5. S. P. Gygi et al., Nat. Biotechnol. 17, 994-999 (1999).
6. X. Yao et al., Anal. Chem. 73, 2836-2842 (2001).
7. J. L. Hsu et al., Anal. Chem. 75, 6843-6852 (2003).
8. A. Thompson et al., Anal. Chem. 75, 1895-1904 (2003).
9. P. L. Ross et al., Mol. Cell. Proteomics 3, 1154-1169 (2004).
10. D. M. Desiderio and M. Kai, Biomed. Mass Spectrom. 10, 471-479 (1983).
11. C. M. Shuford et al., Anal. Chem. 89, 7406-7415 (2017).
12. R. P. Grant and A. N. Hoofnagle, Clin. Chem. 60, 941-944 (2014).
13. L. K. Pino et al., Anal. Chem. 90, 13112-13117 (2018).
14. M. Bantscheff et al., Anal. Bioanal. Chem. 404, 939–965 (2012).

