Advances in Fragment-Based Drug Discovery
Fragment-based drug discovery has established itself as a key approach within the drug discovery field.

Complete the form below to unlock access to ALL audio articles.
Understanding Fragment-Based Drug Discovery. Credit: Technology Networks via YouTube
Over the past decade, fragment-based drug discovery (FBDD) has garnered increasing interest among drug discovery experts and has established itself as a key approach within the field – acting as an alternative starting point for the discovery of high-quality lead candidates. Despite advances in several screening technologies, which have undoubtedly driven FBDD forward, applying a fragment-based approach to more complex biological targets remains a challenge.1
This article discusses the evolution of FBDD, highlights key drug discoveries resulting from fragment-based methods, and explores some of the advantages and challenges of harnessing this approach.
Contents
What is fragment-based drug discovery and why is it useful?
Establishing fragment-based approaches
- Theoretical basis for fragment-based drug discovery
- Detection of fragment binding: Advancing biophysical techniques
- Fragment optimization
- Fragment merging, linking and growing
Drugs derived from fragment-based methods
Strength in numbers: Combining fragment-based methods with other approaches
What is fragment-based drug discovery and why is it useful?
Fragment-based drug discovery starts with a small collection of very small compounds that typically comprise >20 non-hydrogen atoms or "heavy" atoms.2 The size of these compounds means that the number of possible fragments is considerably lower than the number of drug-like molecules typically generated from other drug discovery approaches, such as high-throughput screening (HTS).To put it in to perspective – it is estimated that there are more possible small drug-like molecules than there are stars in the universe!3 In contrast, fragment libraries are composed of just a few thousand miniscule molecules.
Size matters…
The larger the molecule, the more complex it becomes. The greater the molecular complexity the greater probability of interactions with a protein target4 – interactions that may not always be favorable.Small fragments form fewer interactions with a protein target, and therefore you would expect them to be able to more easily bind to more sites on more targets, translating to a greater number of "hits". (Figure 1)
The size of fragment libraries in comparison to to those generated by other approaches, meant that initially fragment techniques were largely pursued by small biotech companies. Prof. Roderick Hubbard, Structural Biology Lab, University of York, UK explains: “The reduced size of the chemical libraries made fragment-based methods accessible to them.”
“It took some time for the majority of medicinal chemists to realize that starting with a small, weak affinity hit is preferable to working with a larger, more potent compound.”

Establishing fragment-based approaches
Prof. György Miklós Keserű, scientific advisor, Medicinal Chemistry Research Group at the RCNS-HAS, Hungary, proposes that: “…there were many important contributions that resulted in the development of the fragment-based drug discovery (FBDD) approach into a validated strategy for both academia and pharma. Concerted efforts by the founders and practitioners of FBDD have solved many problems including; library design, screening and fragment optimization.”Theoretical basis for fragment-based drug discovery
Long before FBDD was physically put in to practice, Jencks published on the theoretical basis for FBDD.6 “The idea of using small pieces of chemistry which are found to bind to the active site of a protein was established in the mid-1980s by the computational- and (subsequently in the early 1990s) structural biology community,” says Hubbard.This theoretical background was further supplemented in 1984 when Andrews et al. proposed that individual functional groups contributed intrinsic binding energies that could be used to determine the ‘goodness of fit’ of a drug to its target.7
Detection of fragment binding: Advancing biophysical techniques
“Fragment-based methods were first practically applied to the drug discovery field by the group of Fesik and Hajduk at Abbott in 1996,”8 comments Hubbard. The Abbott scientists used a nuclear magnetic resonance (NMR)-based method to identify fragments that could bind to proximal subsites of the target protein – fragments that were then optimized and linked to create high-affinity ligands.9 A few years later, Nienaber et al. published a paper on the use of X-ray crystallographic screening for clinical compound discovery, and subsequently, both methods – NMR and X-ray crystallography – became the focus of numerous companies who took on the task of tailoring these strategies specifically to the drug discovery field.“An important and continuing development has been in both the sensitivity and robustness of the various biophysical techniques, used for identifying fragment hits, and importantly, experience in using them,” says Hubbard.
Several other approaches are now adapted for FBDD besides NMR and X-ray crystallography.5
- Substrate activity screening. In this screening strategy, substrates for a specific protein target can be identified and optimized quickly. It allows the conversion of fragments into non-peptidic inhibitors of cysteine and serine proteases.
- SPR spectroscopy. This technique involves immobilizing the target protein (on a metal-coated chip) and allowing fragments to flow past. A change in the fragment-protein mass occurs upon binding which causes a change in the reflective property of the metal chip enabling detection in real-time.
- Virtual screening. Computational approaches can shed light on the molecular interaction between the fragment and protein target. This method can be used to predict structural information and examine the binding of fragments to multiple protein targets.
Fragment optimization
A fragment-based approach is particularly valuable for new targets where more conventional screening (HTS) has already failed. Its ability to identify diverse chemical matter that can bind to the target provides the medicinal chemist with choice when it comes to triaging compounds – allowing them to investigate the most promising fragments with long-term potential for development into drug candidates.A key challenge arises once you have successfully identified the initial hits. You are then faced with the task of transforming these fragments – which possess a weak affinity to the protein target (often with KD values in the high μM to mM range) – into more potent and promising ligands.
“Fragment starting points generally have a ‘good’ physchem profile – they bind to protein hot spots and there is potential to optimize them rationally using structural information and ligand efficiency metrics,” notes Keserű.
Effective fragment optimization often involves two key steps. Firstly, co-structures of the fragments bound to their target proteins are needed to enable you to select the best hits, which can then be considered for development into a lead candidate.7 The second step involves performing several biophysical and biochemical assays, to track the potency and efficacy of the fragment.6 These additional assays complement previously acquired data from the initial biophysical studies, enhancing the predictive power of the medicinal chemist – for rational design.
Fragment merging, linking and growing
There are three main methods used for fragment elaboration. The first is "linking" whereby non-overlapping fragments that bind at different protein target sites are linked together to produce a novel chemical series. Another is "merging", which involves combining the common structural parts of overlapping fragments that complex with the target protein.1 Thirdly is fragment "growing" which involves building "sensible chemistry" around a single fragment hit.10Drugs derived from fragment-based methods
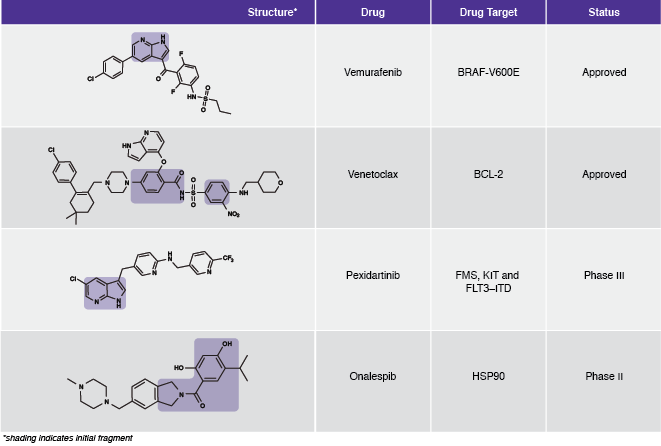
“The successes of the mid-to-late 2000s led to the adoption of fragment-based methods across most companies and there are now many published success stories of advancing from a weak, mM fragment hit to a potent, nM drug candidate for “conventional” drug targets such as kinases and most enzymes,” Hubbard explains.Table 1: Examples of FBDD-derived drugs.
Strength in numbers: Combining fragment-based methods with other approaches
In the early days, FBDD was considered as an alternative approach to HTS. However, more recently, there is a growing consensus that these two methodologies are in fact complementary – rather than competitive – and by adopting both strategies you could be at a distinct advantage.“Although fragment screening was often used as a ‘backup’, for targets without HTS hits, nowadays fragment screening and HTS are applied in parallel. FBDD approaches might help to characterize the druggability of the target and identify structural moieties that are confirmed by HTS. This would contribute to the prioritization and more effective optimization of HTS hits,” notes Keserű.

“Fragments and HTS are a powerful combination. The fragment hits can be used to mine the corporate compound collection for near neighbors which can be assessed in more sensitive assays than can be deployed in HTS. The fragment hits provide many ideas for scaffold replacement in HTS hits,” explains Hubbard, “It remains to be seen how effectively fragments can be combined with other methods such as DNA encoded libraries.”
Fragment-based drug discovery: What does the future hold?
Several technologies have the potential to be developed and improved to maximize the success of fragment-based drug discovery. “I would like to see more sensitive, robust, label-free detection technologies that can be used not only for identifying hit compounds but also supporting optimization in the fragment- to-hit- to-lead stages.” says Hubbard. Advances in screening technologies will be key to driving FBDD forward and to adapting it to more challenging targets. Keserű notes: “There is a remarkable diversification in fragment technologies. SPR and ligand detected NMR is still frequently used, however, X-ray crystallography and protein detected NMR screens are getting more and more popular since these provide direct structural information on fragment binding from the outset.”
Keserű continues: “It is my belief that continuing progress with exploiting computational techniques, supported by high resolution X-ray crystallography and biophysical techniques might help to make the design process more effective.”
Although FBDD has come a long way since the 1980’s there is still a way to go when it comes to learning the best ways to approach more challenging classes of new targets. Hubbard believes that an important consideration “is investing the time to generate the right reagents – both the protein constructs that are needed for biophysical and structural studies, but also in developing robust primary assays to report on ligand binding – including defining just what the target is in terms of post-translational modification or partners that bind to modify function.”
“There remains a tendency to see fragments as a solution when other methods have failed,” concludes Hubbard, “we are still learning the lessons of how to use the techniques on more challenging new classes of targets.”
1. Velvadapu, V., Farmer, BT., Reitz, AB. (2015)
Chapter 7: Fragment-Based Drug Discovery. The Practice of Medicinal Chemistry.
Retrieved from https://www.sciencedirect.com/science/article/pii/B9780124172050000079
2. Erlanson DA, Fesik SW, Hubbard RE, Jahnke W,
Jhoti H. Twenty years on: The impact of fragments on drug discovery. Nat.
Rev. Drug Discov, 2016;15(9),605-619. doi: 10.1038/nrd.2016.109
3. Bohacek RS, Mcmartin C, Guida WC. The art and
practice of structure‐based drug design: A molecular modeling
perspective. Med. Res. Rev. 1996;16(1),3-50. doi: 10.1002/(sici)1098-1128(199601)16:13.3.co;2-d
4. Hann MM, Leach AR, Harper G. Molecular
complexity and its impact on the probability of finding leads for drug
discovery. J Chem Inform Comput Sci. 2001;41(3),856-864. doi: 10.1021/ci000403i
5. Whittaker M, et al. Fragments: past, present and
future. Drug Discov. Today Technol. 2010;7(3),e163-eE171.
doi: 10.1016/j.ddtec.2010.11.007
6. Jencks WP. On the attribution and additivity of
binding energies. PNAS. 1981;78(7),4046-4050. doi: 10.1073/pnas.78.7.4046
7. Andrews PR, Craik DJ, Martin JL. Functional
group contributions to drug-receptor interactions. J. Med. Chem. 1984;27(12)1648-1657.
doi: 10.1021/jm00378a021
8. Shuker SB, Hajduk PJ, Meadows RP, Fesik SW.
Discovering high-affinity ligands for proteins: SAR by NMR. Science. 1996;274(5292),1531-1534.
doi: 10.1126/science.274.5292.1531
9. Hubbard RE. Fragment approaches in
structure-based drug discovery. J Synchrotron Radiat. 2008;15:227-230.
doi: 10.1107/S090904950705666X
10. Scoffin R, Slater M. The virtual elaboration of
fragment ideas: Growing, merging and linking fragments with realistic
chemistry. J. drug discov. dev. deliv. 7(2),36-40.


