Advances in Phenotypic Screening: A Turning Point for Drug Discovery?


Complete the form below to unlock access to ALL audio articles.
Drug discovery approaches can be broadly divided into two categories – target-based and phenotypic screening. Historically, medicines were selected for their visible (or perceived) health benefits, with systematic phenotypic analysis gaining traction in the early 1900s. By the late 1990s, target-based drug discovery dominated the pharmaceutical industry, built on decades of advances in molecular biology.
Today, it is easy to detect dissatisfaction with drug discovery success rates; despite major technological progress and increasing investment in pharmaceutical R&D, there has been a steady decline in the number of new drugs entering clinical development and reaching the market. From 2006–2015, the overall likelihood of progressing from Phase I to U.S. FDA approval was only 9.6 % (based on a BIO report of clinical development success rates, n = 9985). While the shift to target-based drug discovery is not the only explanation for this inefficiency, there is a consensus that we have relied too heavily on assumptions about how well a target or pathway is connected to disease (the amyloid β hypothesis is a classic example).
The choice between target-based and phenotypic drug discovery approaches is not a case of “one or the other”, as reality lies somewhere in the messy middle. However, there is now an urgent need to incorporate research models and technologies that acknowledge the complexity of biological systems. In this article, we explore recent developments related to cell-based phenotypic screening, discuss the elements of a good phenotypic drug screen, and discover how these approaches are helping to provide a more accurate biological context for drug screening.
What is cell-based phenotypic screening?
In the context of drug discovery, cell-based phenotypic assays are compound screening systems focused on the modulation of a cell’s phenotype, in a way that has biological relevance to the disease. While phenotypic screening is largely used for compound screening, it is also used in general research – for example, to study gene knockouts and overexpression, or to compare individual patient samples. In its purest form, phenotypic screening identifies compounds that produce desirable biological effects, without having prior knowledge of their activity or mode of action against a specific target/s. In practice, phenotypic screening projects are not always target-agnostic, says a Senior Computational Biologist at Broad Institute, Dr Beth Cimini: “There’s a spectrum; you can have a target assay that also has a phenotypic component, you can have one that is purely focused on a target, or you can have an assay where you have no target at all.”
Researchers hope phenotypic screening will lead to better predictions of compounds with therapeutic value, as relevant and measurable changes can be observed in a cell-based (or tissue- or whole-organism-based) model that mimics some aspect of disease. Phenotypic drug screening makes use of nuclear dyes, immunohistochemistry, and other tools to observe phenotypic changes such as those related to cell morphology (including nuclei or other organelles), spatial arrangements (patterns and spacing), the production of specific protein/s, gene expression, or cellular death. Sometimes, hallmarks of disease do not correspond to phenotypes that can be easily screened and underlying molecular events are used as surrogates.
What makes a good phenotypic drug screen?
Fundamentally, the most important trait of a good phenotypic drug screen is its translatability, i.e. the ability of the screening assay to predict the clinical therapeutic response to a drug. But what does this mean in practice? How can we know whether a phenotypic drug screen is a good one, before becoming deeply invested in clinical trials? In 2015, Vincent et al. coined the phenotypic screening “rule of 3”, and highlighted three specific criteria to help design the most predictive phenotypic assays:
- System: How relevant is the assay to the disease? Are the cells a suitable choice for the assay?
- Stimulus: How relevant is the stimulus to the disease? Ideally, phenotypic assays would use highly relevant cells, such as patient-derived cells that incorporate disease-causing genetic alterations. Where this is not possible, “best-guess” efforts may be made, such as the use of inflammatory or cytotoxic agents to recreate a cellular injury of interest.
- Endpoint: How well does the assay readout relate to the clinical endpoint? Assays that use a miniaturized version of the clinical endpoint as the assay readout are preferred over those that do not come as close to reproducing key in vivo disease phenotypes.
Patient-derived organoids give hope to personalized medicine
To address the need for more complex cell models that better recapitulate in vivo physiology, a range of microphysiological systems have emerged. In contrast to two-dimensional cell monolayers, microphysiological systems can feature key biological stimuli including mechanical cues (e.g. shear stress and stretch), a three-dimensional microenvironment, sustained concentration gradients, and interactions between multiple cell types. These systems (described as organs-on-a-chip, organoids, 3D spheroids, and static micropatterned technologies) use microengineering techniques, drug delivery and/or sensing tools to examine the effect of drugs and/or the microenvironment on living cells. Human organoids are three-dimensional multicellular in vitro tissue constructs that serve as tools for drug discovery and studies of human-specific disease mechanisms. They are favored for their ability to mimic at least some function of an organ and can be derived from induced pluripotent stem cells or donor tissue.
Intestinal organoids developed from rectal biopsies have been used to study hereditary diseases like cystic fibrosis. In the Clevers laboratory at the Hubrecht Institute in the Netherlands, researchers have been seeking a less invasive approach to organoid development. Using kidney-derived cells found in the urine of people with cystic fibrosis, Schutgens et al. (2019) report the development of primary kidney tubular epithelial organoids known as “tubuloids” which represent proximal and distal nephron segments. In cystic fibrosis, mutations in the CFTR (cystic fibrosis transmembrane conductance regulator) gene compromise the function of ion and fluid transportation across a range of epithelia. To demonstrate the suitability of the tubuloids for ex vivo assessment of treatment efficacy, the group performed the forskolin swelling assay.
Maarten Geurts, a PhD student in the Clevers laboratory, explains how the assay is used to assess drug responses in the organoids: “The addition of Forskolin to healthy intestinal organoids results in a (CFTR-dependent) swelling response within one hour, whereas this response is absent in CF-patient derived organoids. We can test CFTR-restoring drugs by monitoring the Forskolin-induced swelling response after the addition of these drugs to the organoids. As tubuloids show a similar response to Forskolin and can be derived from urine, they can be a less invasive model to test CFTR-restoring drugs in a patient-specific manner.”
Keeping in line with Vincent’s “rule of 3”, there is a strong correlation between restoration of the swelling response by CFTR-restoring drugs and the clinical response of individual patients. Geurts notes that the group gained confidence in the translational relevance of their cell-based phenotypic screening assay when a patient started taking a potentially beneficial drug identified in the laboratory:
“The best way of building confidence in the cell-based phenotypic drug screening assays we perform in our lab is by using potentially relevant drugs directly in patients. We derived organoids from a patient with different mutations in the CFTR gene and showed by the Forskolin-induced swelling assay that Orkambi® (lumacaftor/ivacaftor) resulted in organoid swelling, suggesting a beneficial response in patients. After this patient started taking lumacaftor/ivacaftor, his symptoms reduced significantly, showing that our in vitro phenotypic drug screen could be directly translated to patients.”
Casting the net wider with morphological profiling
For diseases where the desired phenotype is unknown, some believe image-based, high-content screening will provide value, and multimillion-dollar collaborations are being launched to prove it. In contrast to conventional screening assays, which are focused on quantifying a small number of pre-selected features, morphological “profiling” casts a wider net to gather insights based on cell morphology – a potentially rich data source. Following concerns that assays with a single, population-averaged readout may yield poor quality hits, there have been increasing efforts to draw out multiple parameters at the level of single cells and improve image processing, which has traditionally been an extremely time-consuming exercise. Now, microscopes and image analysis can be automated, enabling high-content screening and a less biased approach to profiling.
The Cell Painting assay, co-developed by researchers in the Carpenter and Schreiber Laboratories at the Broad Institute of MIT and Harvard, can yield approximately 1500 morphological features from each stained and imaged cell. The measured features are integrated into a morphological profile and include staining intensities, textural patterns, size, and shape of eight labeled organelles, as well as correlations between stains across channels, and adjacency relationships between cells and among intracellular structures (Figure 1). While most published profiling methods use only three dyes, the Cell Painting assay uses six fluorescent stains imaged in five channels.
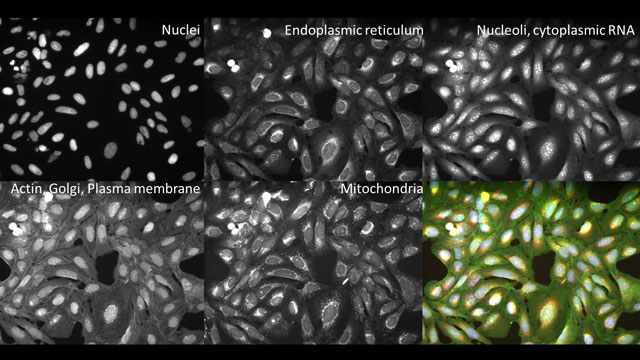 Figure 1. A sample of images featuring specific organelles, extracted from a single Cell Painting assay (bottom right). Image courtesy of Dr Cimini, Broad Institute.
Figure 1. A sample of images featuring specific organelles, extracted from a single Cell Painting assay (bottom right). Image courtesy of Dr Cimini, Broad Institute.Cimini is co-leading development of CellProfiler™, a free and open-source cell image analysis software for morphological feature extraction, and notes that, “the biggest strength of phenotypic profiling is the ability to do it cheaply and easily, and then to be able to create associations.” These associations can then be used to select compounds that would benefit from more focused screening approaches. To this point, Cimini highlights a study where data from a single high-throughput imaging assay was repurposed to predict the biological activity of compounds in other (more expensive or low-throughput) assays, increasing hit rates by 50- to 250-fold.
Are we witnessing a turning point?
As summarized recently by geneticist Sir Adrian Bird, “biology has become one huge canvas” – and phenotypic screening is no exception. The phenotypic screening toolkit continues to expand, from microphysiological systems and morphological profiling, to gene-editing technologies that recreate disease states in vitro, to image-based machine-learning models which predict compound activity based on phenotypic features embedded in the images. Hopefully, we will look back on this time as a turning point, where efforts to extract more biologically relevant information during drug screening translate to improved success rates in the clinic.

