How To Expedite Drug Development by Waiving Certain Clinical Trials

Complete the form below to unlock access to ALL audio articles.
The COVID-19 pandemic has caused numerous obstacles for clinical research across the world. Widespread movement restrictions, a diversion of healthcare personnel to the frontline and a compromised ability to ensure participant safety are examples of the many challenges faced by the pharmaceutical industry. The impact on clinical trials is likely to be long-lasting, creating an urgent need for innovative strategies to ensure the progression of future therapeutic development.
But how can today’s pharma industry make progress and still get products to the market in the face of such long-term disruption? Is it possible to eliminate the need for human trials in some cases?
High-quality in vitro data can accelerate drug development
A critical part of the drug development process is to establish the rate and extent of drug absorption, which are key determinants of a compound’s safety. Adverse effects may occur if a drug is released into the circulation too quickly, while slow or inadequate release may not be sufficient to produce the desired effect. Drug absorption is typically assessed in human studies by measuring bioavailability, which is defined by the US Food and Drug Administration (FDA) in 21 CFR 320.1 as “the rate and extent to which the active ingredient or active moiety is absorbed from a drug product and becomes available at the site of drug action.”
While animal studies can be used to qualitatively categorize drugs into low- and high-bioavailability classes, clinical trials are needed to determine optimal dose concentrations and regimens. Clinical studies are challenging under normal circumstances; they are time consuming, expensive and can be burdensome for subjects. In the current climate, we are witnessing how clinical trial administrators must navigate unique problems posed by the current COVID-19 pandemic, and understand that any opportunities to bypass human studies represent massive time- and cost-savings; we estimate that the replacement of in vivo bioavailability and bioequivalence studies saves $50–150 million per drug product.
Compared to most other routes of administration that bypass intestinal absorption (e.g., intravenous, intramuscular, subcutaneous), the absorption of orally administered products is more complex. For example, first-pass elimination (metabolism in the liver or gastrointestinal tract) can significantly contribute to bioavailability variability and is influenced by food intake. Therefore, circulating drug concentrations must be measured to assess the extent of systemic absorption and metabolism. In human bioavailability studies, two pivotal pharmacokinetic parameters are measured: AUC (area under the curve – an indicator of drug exposure and clearance rate) and Cmax (peak serum concentration). AUC and Cmax are also used to demonstrate bioequivalence, as required during the development of new generic drugs or different formulations.
Fortunately, in the case of orally administered drugs, there are significant opportunities to fast track the development of certain compound classes. By collecting high-quality data from specialized in vitro experiments, it is possible to obtain a “biowaiver” as a surrogate for human bioavailability and bioequivalence studies. The Biopharmaceutics Classification System (BCS) was developed specifically for this purpose, to enable the identification of compounds that meet the criteria for a BCS-based biowaiver. This system works by classifying drugs on the basis of the three determinants of drug absorption for immediate-release solid oral dosage forms:
- Dissolution: how quickly the compound dissolves to form a solution
- Aqueous solubility: the maximum concentration of a solute that can dissolve at a given temperature
- Intestinal permeability: the extent to which the drug is absorbed across the gastrointestinal wall
The BCS can thus aid drug developers as they seek innovative solutions to help them progress drug development amid current industry challenges.
Biowaivers bring more benefits than meets the eye
By leveraging biowaivers, developers will be better able to weather the disruption that looks to be staying with us for the foreseeable future. Limiting unnecessary research involving human subjects not only benefits drug developers and regulatory bodies through time- and cost-savings; shorter development timeframes ultimately benefit patients too. Replacing human bioequivalence studies with in vivo experiments may allow drugs to become available that would have otherwise been thwarted or delayed by the current barriers to clinical trials, providing patients with earlier access to new therapeutics.
In vitro bioavailability studies are also preferred for certain compounds that carry a higher risk to trial participants, such as those with addictive properties. The collection of data from rigorous in vitro experiments can also lead to better success in later clinical trials. For example, complex drugs with highly variable active pharmaceutical ingredients (APIs) are more vulnerable to in vivo bioequivalence failures, and therefore the acquisition of robust in vitro data helps accommodate variability in drug behavior.
What to expect: the stages of a BCS-based biowaiver application
The FDA-specified criteria for biowaiver eligibility are based on observations related to drug dissolution, solubility and absorption. Differences in drug dissolution can cause the rate and extent of drug absorption to differ markedly across pharmaceutically equivalent solid oral products. For solid oral drugs with rapid dissolution and high solubility, drug absorption is unlikely to be dependent on drug dissolution in vivo or gastrointestinal transit time. Provided the inactive ingredients used in the dosage form do not significantly affect the absorption of APIs, the demonstration of bioavailability or bioequivalence in human studies may, therefore, not be necessary.
To obtain a biowaiver, the drug compound in question must be assessed and categorized into one of the following BCS classes:
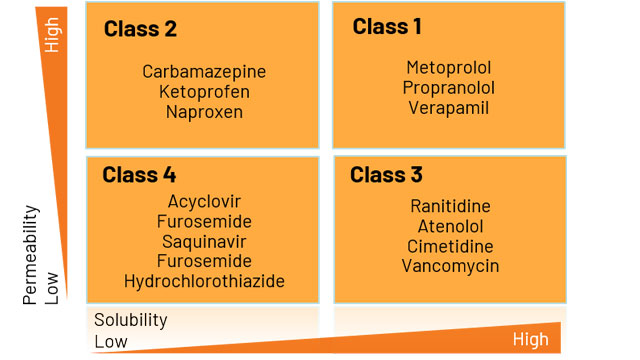
Figure 1: Diagram of BCS classification categories based on permeability and solubility.
Compounds categorized as Class 1 or 3 are eligible for BCS biowaivers. The FDA has published details on recommended test methods for BCS classification, as well as specific parameters that can be used to define whether a drug substance is highly soluble, highly permeable, or rapidly dissolving.
The process of biowaiver applications can be divided into four main stages:
1) A prequalification step to determine eligibility
We recommend the use of selected, early experiments to provide a “go or no-go” decision point, ensuring that the compounds put through to the next stage are those with a high chance of meeting the FDA’s criteria. At this stage, there are two main questions that need to be addressed:
- Is permeability greater than that of our internal standard?
- Is the efflux ratio < 2?
With the right expertise, the prequalification step can take 1–2 weeks and is an opportunity to gain insights into the properties of the drug substance, prior to classification. The prequalification step is also used to establish the system’s suitability for the drug substance in question, as the use of in vitro permeability tests is only recommended for drugs transported by passive mechanisms (i.e. those with an efflux ratio < 2).
2) Optimize protocols for FDA-mandated experiments
Protocols are optimized to ensure reproducible permeability data can be obtained under Good Laboratory Practice conditions, using the most cost-effective options available.
3) BCS classification
FDA-mandated experiments are conducted to obtain permeability, solubility and dissolution data using the approaches discussed below.
4) Respond to questions from the FDA
The final stage of BCS biowaiver applications involves responding to queries from the FDA to ensure they are satisfied with the data provided.
Tools and approaches for biopharmaceutics classification
A successful BCS study design considers which physiological processes are being modeled and includes the use of appropriate internal standards. For in vitro permeability tests in particular, internal standards can play a significant role in determining the success of biowaiver applications. The High Permeability Internal Standard (HPIS) sets the lower threshold of permeability (Papp) for in vitro tests, in which the compound of interest is compared against the HPIS. To be classified as highly permeable, the permeability of the drug compound must be no less than that of the HPIS.
However, the most commonly used HPISs have a relatively high Papp across Caco-2 cell monolayers (human colon adenocarcinoma cells, the most widely used approach to assessing intestinal permeability), increasing the risk of type II errors when predicting in vivo permeability. An optimal HPIS, with a lower Papp, is recommended for in vitro permeability studies.
To increase robustness and throughput, multiplexed systems allow multiple dose forms and/or multiple parameters to be assessed at once. A novel system has also been developed to enable the simultaneous measurement of dissolution of a drug product and absorption of the API across Caco-2 cell monolayers, which are considered the gold standard for in vitro prediction of intestinal permeability. Solubility is assessed separately, typically using the standard shake-flask system.
A strategic approach boosts biowaiver application success
Under the challenging circumstances of the current COVID-19 pandemic, innovative approaches are needed to maximize opportunities for advancing drug development. Fortunately, through a unique classification system, it is possible to gain exemptions from human bioavailability and bioequivalence studies for certain drug classes. This allows drug development companies to move forward while reducing the need for human studies. As a result, patients can gain access to drugs that might not have been able to progress otherwise.
The BCS was developed by the FDA to provide a strategic, scientific framework for the classification of novel and generic compounds, according to their solubility, dissolution and permeability characteristics. This system enables development timeframes to be shortened for compounds categorized as Class 1 (high solubility, high permeability) and Class 3 (high solubility, low permeability), and significant efforts have been directed at improving the chances of biowaiver success. Applications can benefit from the inclusion of a strategic preliminary screening step, well-established methods and an optimal internal permeability standard, and guidance from experienced biowaiver application specialists.
Written by Chris Bode, Ph.D. VP of Scientific and Corporate Communications at Absorption Systems.

