5 Key Steps to Successful CRISPRi

Genome editing technologies are revolutionizing the life sciences, and CRISPR technology is a key player that regularly updates its multifunctional platform. In select bacteria and archaea, CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) and CRISPR associated systems (Cas) target and cleave foreign DNA elements via base-pair forming RNAs, to confer adaptive immunity. The mechanism is experimentally used (and optimized) in labs within a host-independent environment, to target and edit a specific genetic code or reprogram the genome to manipulate cells. While diverse CRISPR systems exist in various organisms, the simplest version is the type II CRISPR, consisting of a Cas9 catalytic enzyme and two RNAs; a mature CRISPR RNA (crRNA) and a trans-acting RNA (tracrRNA). Researchers combined the two-component native RNA system to engineer a chimeric single guide RNA (sgRNA) in the lab, enhancing experimental efficacy. In 2012, a team of scientists produced a further variation to the CRISPR system by integrating a nuclease deficient, mutant Cas9 or "dead Cas9" (dCas9), to repurpose the system and regulate a DNA sequence for its alteration and repression without cleavage/editing, creating CRISPR interference (CRISPRi).
CRISPRi assimilates a catalytically dead Cas9 lacking endonuclease activity for coexpression with a guide RNA (gRNA). The dCas9:sgRNA machinery then forms a DNA recognition complex that can either strand-specifically interfere with transcription elongation or bind to sites of RNA polymerase (RNAP) to prevent transcription initiation, blocking the production of messenger RNAs (mRNAs) overall. Base-pair formation between the RNA chimera and the target DNA is enabled by a short DNA sequence located at the 3’end of the target sequence, known as the "protospacer adjacent motif" (PAM) (Figure 1). The inactive dCas9 mutant is engineered via two point-mutations in the RuvC-like (D10A) and HNH nuclease (H840A) domains. The dCas9:sgRNA machinery can be fused with additional effector domains for effective gene interference (CRISPRi) or gene activation (CRISPRa).
Key highlights of CRISPRi (including pros and cons)
- The machinery is defined by an inactive dCas9 protein repurposed for targeted genome regulation.
- The dCas9: sgRNA machinery allows complementary base-pair formation via a 20-base pair region of the sgRNA, to target specific genomic sites on the DNA sequence of interest.
- CRISPRi can regulate multiple genes with multiple RNAs without off-target effects.
- The silencing is inducible and reversible, while highly specific in bacteria.
- In mammalian cells, transcription repression is comparatively variable and modest.
- The requisite PAM sequence on the foreign DNA of interest may limit the binding sites.
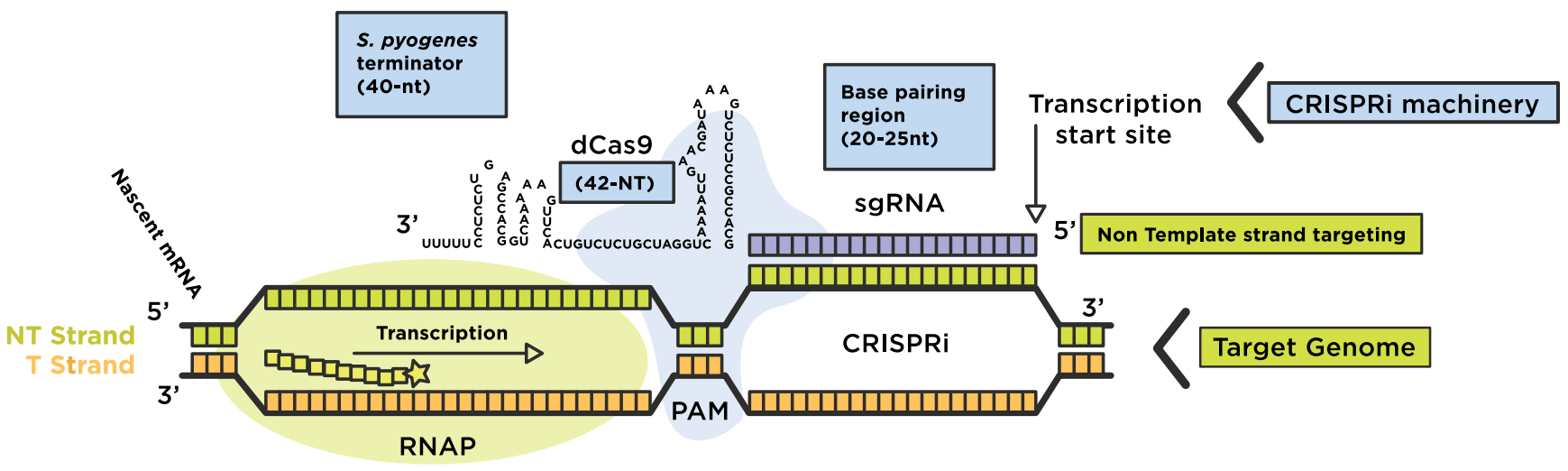
Figure 1: Repurposing the CRISPRi system to regulate a genome. The dCas9-sgRNA complex forms base-pairs with the non-template strand of the target DNA region of interest, for transcription interference (elongation or initiation). Original image adapted from Addgene. Source: Lei S Qi (Stanford University)
The CRISPRi machinery presents an efficient, reversible and specific genome regulating platform for transcription control, without altering the target DNA sequence. The following guideline includes key steps on implementing CRISPRi using dCas9 derived from Streptococcus pyogenes.
Would you prefer to read this as a PDF?
DOWNLOAD HERE
Plan the CRISPRi Experiment - Experimental Design
Once you decide on the desired genetic manipulation, use the following variables to design the experimental framework for CRISPRi in mammalian cells and bacterial organisms.
- Select the species of Cas9, design the dead Cas9 (dCas9).
- Design the sgRNA sequence for site-specific base-pairing, with minimal off-target effects.
- Select the expression system for Cas9 and sgRNA.
- Choose a selectable marker (drug or fluorophore to quantify)
- Select the delivery method.
- Select the detection method.
Select the target site for CRISPRi
The binding specificity of the dCas9-sgRNA complex to the target DNA depends on an NGG PAM motif. For flexible target site-specific selection within a genome, CRISPRi targeting is via straightforward Watson Crick base-pairing between the complementary 20-base pair (bp) region of sgRNA and the target DNA sequence, enabled via the essential 3-nt PAM sequence.
- Additional Cas proteins from variant species and their corresponding PAM sequences are available at Addgene.org.
- The chimeric sgRNA contains a 20-nucleotide (nt) base-pairing region 2) a dCas9 handle hairpin (42-nt) and 3) an S. pyogenes derived terminator (40-nt) to mimic the natural RNA complex.
- Design the 20-nt sgRNA sequence to target the promoter/enhancer region of your gene of interest, or to the beginning of the coding sequence in the 5’ end of the target gene.
- Use the Basic Local Alignment Search Tool (BLAST) to check the specificity of sgRNA binding in the genome. To rule-out additional binding sites and off-target effects, BLAST search the 14-nt specificity region, which includes the 12-nt ‘seed region’ of the sgRNA and 2-nt of the 3-nt PAM region, in the genome of interest.
- For genome mapping of large numbers of short nucleotide sgRNAs use SeqMap to compute off-target effects. Use only sgRNAs without predictable off-targets.
- Append the dCas9 handle and S. pyogenes terminator sequence to the 3’ end of the base-pairing region, predict the chimeric sgRNA secondary structure using Quickfold or RNAfold simulation algorithms.
- Confirm that the sgRNA sequence does not contain sequences for restriction enzyme sites (e.g. EcoRI, Bg1II and BamHI for bacterial sgRNAs; BstXI and XhoI for human sgRNAs), prior to cloning the templates into an expression vector.
- Optional step: to regulate the repression efficiency, introduce single or multiple mismatches into the sgRNA base-pairing region.
Select the expression systems for Cas9 and sgRNA
In mammalian cells, the catalytically dead Cas9 (dCas9) can be fused to a transcriptional repressor domain such as KRAB (Krüppel associated box), for enhanced repression. Conversely, to activate gene expression use an activation domain such as VP16 or VP64, paired to dCas9 in mammalian cells. To functionalize CRISPRi, you require both dCas9 and gRNA templates expressed in your target cells via expression vectors.
- Choose the appropriate expression vector for sgRNA and dCas9. Major expression systems and variants for use in mammalian cells are available online.
- For plasmid or vector-based expression systems of CRISPRi in mammalian systems and in bacteria browse the available constructs.
- The following plasmids reposited by the Stanley Qi Lab, are recommended for dCas9 and sgRNA expression in bacterial organisms and mammalian cells.
- If the plasmid you’re using does not co-express gRNA and dCas9, use separate expression vectors to target repression at the specific locus of interest, as indicated above.
- When cloning chimeric sgRNAs into a small expression vector (5 kb), use oligo PCR and digestion/ligation methods to avoid PCR errors that could be introduced from iPCR.
- To clone multiple sgRNAs into a single expression vector, use the BioBrick assembly method or Golden Gate cloning procedure to multiplex CRISPRi.
- The vectors can be transformed into a cloning strain of E.coli such as the commercially available One Shot Top10 Chemically competent E.coli cells, and then grown to a sufficient quantity for plasmid purification.
- The expression systems should notably include selection markers (e.g. Neomycin) or reporter genes (e.g. GFP) to validate the genetic modification after delivery to target cells.
- A negative control for sgRNA expression is an expression vector without the 20-nt base-pairing region. A negative control for dCas9 expression is the same expression vector without the dCas9 coding sequence.
Deliver the dCas9 and sgRNA
I use IRDye conjugated antibodies from LI-COR, which can be detected by their Odyssey® Imaging Systems. These dyes can be detected in either the 700nm or 800nm channels.
- For targeted gene expression in bacterial systems: co-transform the sgRNA vector, with an inducible dCas9 expressing vector (e.g. #44249) into the desired E.coli strain (e.g. MG1655).
- For targeted gene repression in mammalian cells (e.g. HEK 293 cells – human embryonic kidney cell lines), culture cell lines in supplemented growth media prior to transfection.
- Transfect the mammalian cells using commercially available DNA transfection reagent, follow the manufacturers’ protocol.
- Transfect the dCas9 expression plasmid and the sgRNA expression plasmid per cell-culture well.
- For mammalian cells, measure the gene expression 72 hours after transfection.
Design functional validation assays
Once the gRNA and dCas9 are successfully delivered to your target cells it is time to validate the expected CRISPRi regulation or the level of genome repression.
- If the target gene is fused to a fluorescent protein or LacZ; use either flow cytometry or the measurement of β-galactosidase activity as functional assays to measure the effects on protein expression.
- Functional assays to measure transcription repression of the endogenous genes can also include qRT-PCR or genome-wide NET-Seq technologies.
- For instance, Net-Seq can indicate the exact genomic loci being disrupted via the dCas9: sgRNA machinery’s interference.
- Since the efficacy of CRISPRi can vary between cell types and growth conditions, perform all assays in the same cell type with similar growth conditions.
- The summarized guidelines listed here describe CRISPRi (sgRNA and dCas9 expression to repress a target gene of interest) in E.coli (e.g. strain MG1655) or in human cells (e.g. HEK 293 cells).
- In previous studies, the dCas9 and sgRNAs have facilitated robust silencing and gene repression with higher efficiency in bacteria (99-99.5%), compared to moderate levels in human cells (~50-60%), with possibility for optimization.
References
- Larson M.; Gilbert L.; Wang X.; Lim W.; Weissman J.; CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat Protoc. 2013; 8 (11), 2196.
- Weiyue J.; et al. Specific Gene Repression by CRISPRi System Transferred through Bacterial Conjugation. American Chemical Society, Synthetic Biology. 2014; 3 (12), 931.
- Qi, L.; Larson M.; Gilbert L.; Doudna J.; Weissman J.; Arkin A.; Lim W.; Repurposing CRISPR as an RNA-guided Platform for Sequence-Specific Control of Gene Expression. Cell. 2013; 152, 1183.
- Jinek M.; et al. A programmable dual-RNA-guided DNA endonucelase in adaptive bacterial immunity. Science. 2012; 337, 821.
- Inoue H.; Nojima H.; Okayama H.; High efficiency transformation of Escherichia coli with plasmids. Gene 1990; 96:23-28.
- Roy K.; Smith J.; Vonesch S.; Lin G.; Tu C.; Lederer A.; Chu A.; Suresh S.; Nguyen M; Tripathi A.; et al, Multiplexed precision genome editing with trackable genomic barcode in yeast. Nature Biotechnology, 2018; 1696
- Charpentier E. and Doudna J. Rewriting a genome.Nature, News and Views 2013; 495, 51
- Ran F.; Hsu P.; Wright J.; Agarwala V.; Scott D.; Zhang F. Genome engineering using the CRISPR-Cas9 system. Nature Protocols, 2013; 8 (11) 2281
- Myers S.; Wright J.; Peckner R.; Kalish B.; Zhang F.; Carr S., Discovery of proteins associated with a predefined genomic locus via dCas9-APEX-mediated proximity labeling. Nature Methods, Brief Communication, 2018; 5.
- Hannon, G.J., RNA interference. Nature, 2002; 418, 251.
- Beerli, R.R., and Barbas, C.F., Engineering polydactyl zinc-finger transcription factors. Nat. Biotechnol. 2002; 20, 141.
- Wang, H.H., Isaacs, F.J., Carr, P.A., Sun, Z.Z., Xu, G., Forest, C.R., and Church, G.M. Programming cells by multiplex genome engineering and accelerated evolution. Nature, 2009; 460, 898.
- Ledford H., Riding the CRISPR Wave, Nature, News Feature, 2016; 531, 159




