Advances in High-Throughput Screening


Complete the form below to unlock access to ALL audio articles.
Ever since the advent of the multi-well plate, researchers have been pioneering new ways to miniaturize assays for high-throughput screening (HTS). Since then, advances in robotics and liquid-handling devices have made it easier than ever to carry out millions of screens simultaneously. Meanwhile advances in gene editing such as CRISPR, bioengineering and high content imaging mean that HTS is no longer limited to two-dimensional layers of cells in a multi-well dish. Today’s screens can be carried out using engineered pluripotent stem cells that mimic whole organs such as the liver,1 bioengineered surfaces that recreate the tumor microenvironment – so-called “cancer on a chip”,2 and even whole organisms such as zebrafish3 for toxicology screens. Increasingly, the lines are being blurred between high-content and high-throughput and it is computers, not humans, that are spotting the hits. Here, we share two examples that illustrate the power of modern HTS in drug discovery.
Combining computational and in vitro HTS
Professor Natasha Kirienko at Rice University uses a small worm called C. elegans for much of her work on screening for new cancer drugs. Her lab is focusing on drugs that target the mitochondria in cancer cells. Much of the work uses C. elegans because it has many benefits for HTS. “When we want to screen thousands of compounds, we use C. elegans because it’s much faster. Worms are more robust that cultured cells, they are transparent so it’s easier to observe fluorescence, you can use an exact number in each well, and we are doing screening in the context of the whole animal. With worms, we know that if a drug is too toxic for any tissues, the worms will die and the hit will be eliminated from screening. It means we’re already testing for bioavailability and decreased toxicity, so we can eliminate false positives at the very initial stages of screening.
Professor Kirienko’s lab is using this approach to screen molecules to identify novel compounds that can target mitochondria. “It is much easier to screen this high number of molecules in C. elegans and then take those hits and apply the hits to cell lines for more careful evaluation and prioritization.”
Recently her lab published results from a study where they started with computational modeling4 before conducting a more targeted screen.
“Many of the commonly used chemotherapeutics for leukemia target mitochondria as well as replicative processes. In other cancers, we see less of these kinds of drugs. We wanted to see if there was an unbiased way to predict which type of therapy would be effective for which kind of cancer.”
They started by analyzing a set of about 200 of US Food and Drug Administration (FDA) approved drugs for which some mechanism of action is known. From these, they selected 14 that are known to target mitochondria and 14 that do not. They then retrieved the available sensitivity scores for each cell line from the NCI-60 Human Tumor Cell Lines Screen, computed the scores for mitochondria-targeting drugs and for control drugs for six different cancer cell lines and then ranked the sensitivities.
“After ranking, we noticed that the distribution was not normal. For example, there were only six leukemia cell lines and all of them were at the very top of the list. Further analysis showed that this couldn’t be random. Indeed, when we analyzed all possible permutations of the six cell lines, we found that leukemias are significantly sensitive to mitochondrial damage. At the other end of the spectrum, we had ovarian cancers which were not. Moreover, this polar distribution was only for mitochondria targeting drugs i.e. we find a significant association when we look at sensitivity to mitochondrial drugs and type of cancer, but not sensitivity of non-mitochondrial targeting drugs and type of cancer.”
When they validated this experimentally, as predicted – leukemias were sensitive to multiple kinds of mitochondrial damage, whereas other cancers and control healthy blood cells were much less sensitive. Moreover, when they added an inhibitor of glycolysis to target the cell’s ability to make energy and maintain electron flow, they found a synergistic effect. Now they are testing multiple drug combinations where drugs currently used in clinics that target mitochondria are being combined with other treatments that cause mitochondrial damage. They expect these results to be published soon.
Ligand-Receptor Binding Kinetics on the PHERAstar® Microplate Reader
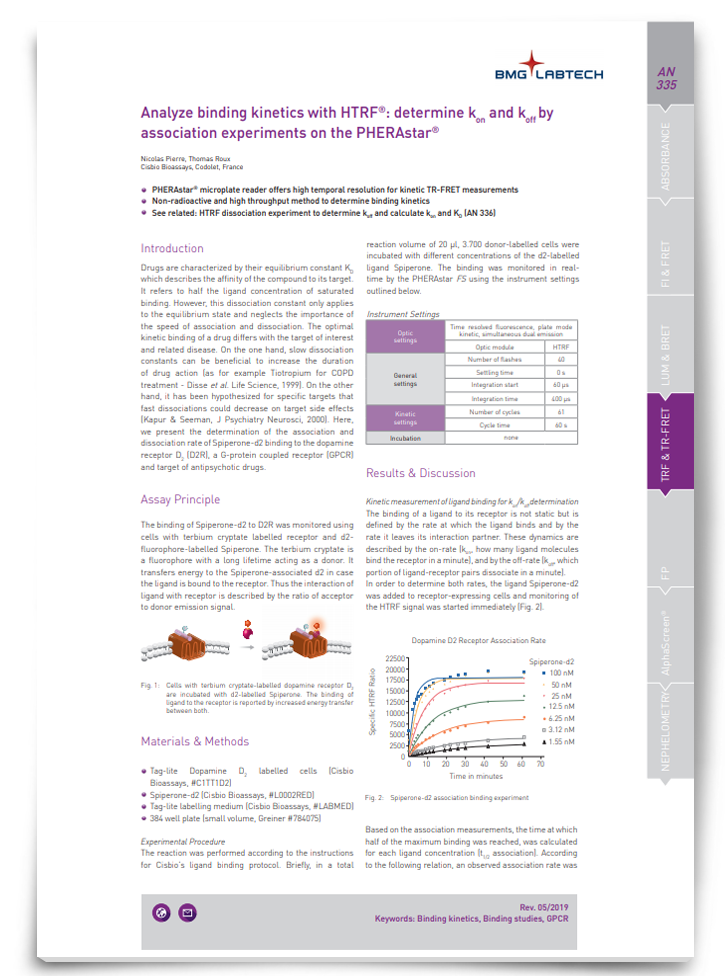
Drugs are characterized by their equilibrium constant KD, which describes the affinity of the compound to its target. However, this dissociation constant only applies to the equilibrium state and neglects the importance of the speed of association and dissociation. In this app note, discover a method for analyzing binding kinetics.
Download App NoteSponsored Content
Image-based epigenetic HTS
Phenotypic screening has become the tool of choice for drug discovery. However, there is a lack of methods for effective and efficient screening for epigenetic-targeting drugs, says Professor Alexey Terskikh at Sanford Burnham Prebys Medical Discovery Institute. His lab has developed a new approach – Microscopic Imaging of Epigenetic Landscapes (MIEL).5
“My lab has been studying neurogenesis for a long time. Then, I was struck by presentations I saw at conferences where patterns of DNA methylation appeared differently in pluripotent cells and neuroprogenitors. At another conference I heard about machine learning approaches to extract information from these patterns. However, it still took us quite a while to realize how powerful the approach could be.”
“People were starting to question the results of phenotypic screens using reporter or antibody assays because they pick up patterns that are pretty much arbitrary. But when we use epigenetic markers, there’s not more uncertified patterns – we’re looking at the epigenome of the cell.”
This prompted Terskikh and colleagues to start to look at DNA methylation, acetylation – or any marker that is related to the chromatin of the cell. The MIEL platform captures the nuclear staining patterns of different epigenetic markers and uses machine learning to distinguish between the patterns.
“We’re looking at three-dimensional (3D) patterns now – voxels instead of pixels. When we use 3D arrangements it becomes even more apparent that that the epigenome has a certain structure. That structure is in fact the identity of the cell. If I can perfectly determine the volume and distribution of a certain epigenetic mark and I find a different cell that has the same pattern, especially at high resolution, I can guarantee that those are the same cells. The resolution is not perfect yet, of course, but within the realm of phenotypic screening it’s probably as close as you can get.”
Targeting the cGAS-STING Pathway Using a Homogenous, HTS Compatible cGAS Activity Assay

Cyclic GMP-AMP synthase (cGAS) is a recently discovered enzyme that acts as a foreign DNA sensor to elicit an immune response to pathogens via activation of the STING (stimulator of interferon genes) receptor. Since its discovery, aberrant activation of cGAS by self-DNA has been shown to underlie some autoimmune diseases, such as systemic lupus erythematosus (SLE) and Aicardi–Goutieres Syndrome (AGS).
View PosterSponsored Content
Although a phenotypic screening method, MIEL is geared towards high throughput. All the experiments have been done in a 384-well plate and the lab is moving to a 1536-well plate format that could screen as many as half a million compounds. “This means you can now combine the beauty of phenotypic screening which is low throughput with analysis of patterns of the epigenetic meaning of that screen which is high throughput.” The great advantage over conventional epigenetic screens, Terskikh says, is that it moves away from the threshold approach of reporter-based assays where you are looking for an up- or down-regulated signal. “There is no question that you won’t detect epigenetic marks. These marks are always present. We’re talking about a pattern that corresponds to the cell, not the presence of absence of a signal.”
Terskikh plans to use the approach to screen for pro-longevity drugs that can move the needle on the epigenetic clock. “We’ve shown that we can use MIEL to correlate epigenetic patterns with the age of cells and we’re getting better and better at it. Now we want to see if we can use this method as a cell-based screen for anti-aging compounds. Only 10–15 such drugs have been discovered to date and they have all been through serendipity, so if one has a tool to screen through thousands of compounds and find new pro-longevity drugs, that’s really exciting.”
References
1. Corbett JL and Duncan SA. (2019) iPSC-Derived Hepatocytes as a Platform for Disease Modeling and Drug Discovery. Front Med (Lausanne). DOI: 10.3389/fmed.2019.00265.
2. Fetah KL et al. (2019) Cancer Modeling-on-a-Chip with Future Artificial Intelligence Integration. Small. DOI: 10.1002/smll.201901985
3. Jia HR et al. (2019) Nanomaterials meet zebrafish: Toxicity evaluation and drug delivery applications. J Control Release. DOI: 10.1016/j.jconrel.2019.08.022. Epub 2019 Aug 22. Review. https://www.ncbi.nlm.nih.gov/pubmed/31446084
4. Panina SB et al. (2019) A mechanism for increased sensitivity of acute myeloid leukemia to mitotoxic drugs. Cell Death Dis. DOI: 10.1038/s41419-019-1851-3
5. Farhy C et al. (2019) Improving drug discovery using image-based multiparametric analysis of the epigenetic landscape. eLife. DOI: 10.7554/eLife.49683

