Using Single Cells To Get the Whole Picture of the Epigenome

Complete the form below to unlock access to ALL audio articles.
Genes may determine what characteristics are passed down from parent to offspring, but each cell expresses these genes differently based on “external” epigenetic modifications. Epigenetics don’t alter the gene sequence (genotype), but they do influence cell behavior and function (phenotype). The study of epigenetics helps us understand how phenotypic changes lead to disease, stem cell differentiation, and essentially, what drives the fate of every cell in the human body.
The epigenome is not consistent between cells, or even between cells of the same type. Individual modifications come and go throughout a cell’s lifetime. Therefore, scientists are faced with the steep challenge as they try to decipher the role of epigenetics in disease and development.[i] Understanding intercellular heterogeneity is key here. The epigenome must be examined at single-cell resolution.
Now, with the advancement of single-cell sequencing methods like the single-cell assay for transposase accessible chromatin (scATAC-seq), researchers have access to sophisticated techniques to map large cell populations, one cell at a time. The resulting epigenomic information provides unprecedented insight into the different cell types that come together to form organs and organ systems, as well as pathogenic modifications associated with disease.
A map of the epigenome to understand cell differentiation
When genetic information is packed tightly in the form of inaccessible chromatin (heterochromatin), it is poorly expressed because transcription machinery can’t reach it. Epigenetic modifications of DNA control chromatin packing within a cell, dictating how much or little of a gene is expressed and, subsequently, that specific cell’s phenotype.
Every single cell has unique epigenomic instructions that guide how it expresses its genes – and these instructions are subject to change. A map locating epigenetic modifications in the genome would help scientists understand how epigenetics drives cellular differentiation. But until recently, epigenetic assays mainly focused on select regions of DNA or gave bulk results across an entire sample of cells.[ii] These assays were not designed to detect epigenetic patterns in individual cells.
Single-cell tools like scATAC-seq help us get a grasp on intracellular heterogeneity, differentiate between cell populations and map the role of epigenetics in the larger context of an organism. By building a collection of scATAC-seq data, scientists have begun generating a “cell atlas” to provide insight into the role of epigenetics during the intricate biological processes that occur throughout the human lifetime.
The evolution of scATAC-seq
scATAC-seq was built on an earlier method to study open chromatin, nucleosome positioning and transcription factor occupancy, called ATAC-seq.[iii] A single ATAC-seq assay can process hundreds to thousands of cells in a bulk reaction to provide an average of the epigenetic state of the sample population.
During ATAC-seq, a hyperactive transposase mutant, Tn5, binds to open chromatin (euchromatin) regions. Wherever Tn5 binds, it cleaves the DNA and attaches sequencing adapters. Then, after PCR amplification, ATAC fragments are sequenced to identify open chromatin regions. ATAC results indicate where nucleosomes are typically positioned in the cell sample and which regions of the genome are open for transcription factors to bind. As such, scientists use ATAC-Seq as a first-pass screening approach to identify changes in chromatin accessibility between samples.
ATAC-seq has many practical applications, but it can’t account for the cell-to-cell variability that’s often an important aspect of developmental processes and disease. So, researchers developed a new assay in which microfluidic technology is used to isolate individual cells before ATAC-seq.[iv] This assay provides epigenomic information at single cell resolution, earning it the name scATAC-seq.
The key to the scATAC-seq method is that it isolates genomes of individual cells early on to perform a separate ATAC-seq reaction on each individual cell. Then, open regions of the genome are cleaved by the Tn5 transposase, tagged with sequencing adapters and amplified with barcoded cell-identifying primers. Subsequently, the barcoded libraries of ATAC fragments, (each representing an individual cell) are pooled together and sequenced to reveal open chromatin regions of thousands of individual cells.
Charting unknown territory with scATAC-seq
scATAC-seq has proved itself a valuable technique to understand how human diseases like cancer and autoimmune disorders originate in the body. But it is still limited by the number of cells it can process in one go. To address this problem, scATAC-seq has now been refined into two methods that can each assay more cells per experiment through a more scalable, droplet-based technology to isolate individual nuclei (Figure 1).
The first droplet-based iteration of the scATAC-seq method (dscATAC-seq) uses a single cell isolator to encapsulate thousands of individual nuclei in nanoliter-sized droplets for ATAC sequencing. It uses a custom Tn5 transposase to enhance library complexity and signal resolution. Compared to the original microfluidic method, the new workflow is faster and yields greater biological insight with less time and effort spent on sequencing. To demonstrate its power and potential, this technique has been used to conduct an unbiased analysis of the many different cell types and regulatory elements in a mouse brain. [v]
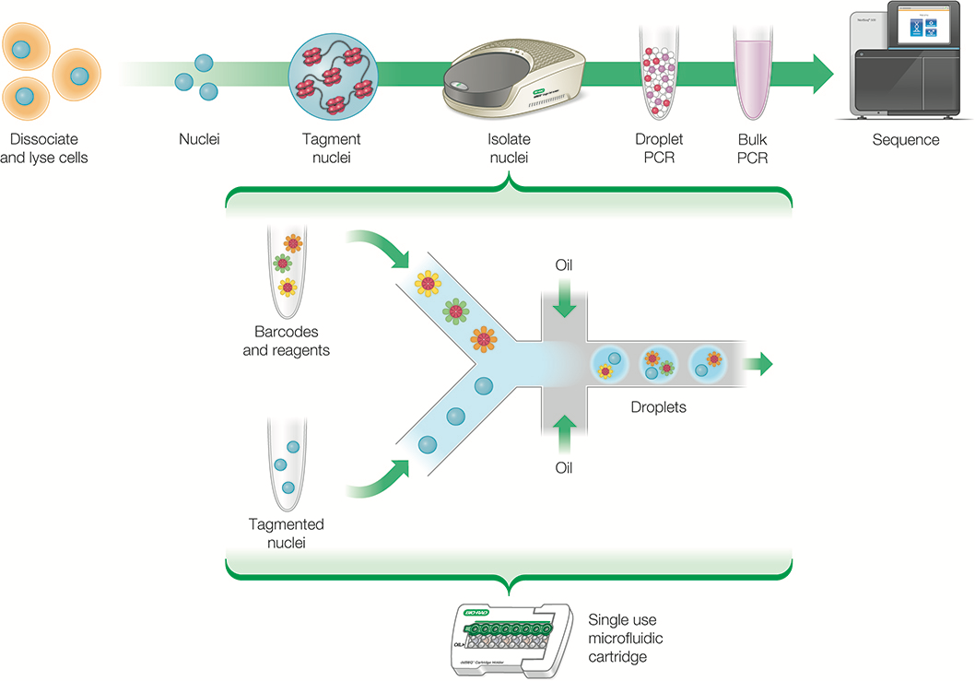
Figure 1: In scATAC-seq, droplet-based technology partitions thousands of whole cells or nuclei into individual nanoliter-sized droplets, enabling researchers to prepare a library of ATAC fragments for sequencing to reveal open chromatin regions. Credit: Bio-Rad Laboratories.
To capture single cell data on a truly massive scale, combinatorial indexing was next introduced into the dscATAC-seq workflow. This new method, called dsciATAC-seq, enables researchers to assess up to 50,000 cells in a single assay. Assaying a large volume of cells is possible because, in dsciATAC-seq, the hyperactive mutant transposase integrates a first set of barcodes as it cleaves open regions of chromatin in each nucleus. Because every cell’s DNA already carries a barcode, multiple cells can be loaded into a single droplet. Then, as usual, ATAC fragments are amplified with a second set of barcoded primers. After sequencing these fragments, the two sets of barcodes are used to derive epigenomic profiles for tens of thousands of cells.
Putting the dsciATAC-seq method to the test, researchers have studied immune cell clusters from human bone marrow derived cells to illustrate how the chromatin accessibility landscape in these cells changes according to different stimulants at the single cell level.5
Although the number of cells that a single scATAC-seq experiment can evaluate has grown dramatically, it will take a continued concerted effort from scientists across many disciplines to create a comprehensive map of the human epigenome, encompassing data from trillions of cells.[vi] Furthermore, to help decode the patterns we find in the human epigenome, it may be valuable to gather information about the epigenomes of animals commonly used as research models. As each of these maps become increasingly detailed, scientists will gain a more thorough understanding of how biological process work and may apply this knowledge towards developing better treatments for complex diseases.
Reference:
[i] Egger, G., et al. Epigenetics in human disease and prospects for epigenetic therapy. Nature, 2004, 429, 457–463. doi:10.1038/nature02625
[ii] DeAngelis, J. T., Farrington, W. J., & Tollefsbol, T. O. An overview of epigenetic assays. Molecular biotechnology, 2008, 38(2), 179–183. doi:10.1007/s12033-007-9010-y
[iii] Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature Methods, 2013, 10(12):1213-8. doi: 10.1038/nmeth.2688.
[iv] Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY, Greenleaf WJ. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature, 2015, 523(7561):486-90. doi: 10.1038/nature14590.
[v] Lareau, C.A., Duarte, F.M., Chew, J.G. et al. Droplet-based combinatorial indexing for massive-scale single-cell chromatin accessibility. Nature Biotechnology 37, 916–924 (2019) doi:10.1038/s41587-019-0147-6.
[vi] Bianconi, E., Piovesan, A., Facchin F., Beraudi, A., Casadei. R., Frabetti, F., Vitale, L., Pelleri, M., Tassani. S., Piva, F., Perez-Amodio, S, Strippoli, P. & Canaider, S. An estimation of the number of cells in the human body. Annals of Human Biology, 2013, 40:6, 463-471. doi: 10.3109/03014460.2013.807878.

