Troubleshooting the Development of New Gene Therapies

Complete the form below to unlock access to ALL audio articles.
Gene therapy does more than treat genetic diseases – it can cure them. A one-time dose of a non-replicative viral vector, such as commonly used recombinant adeno-associated virus (AAV), delivers a functional gene to replace or compensate for a dysfunctional version that is causing a patient’s disease (Figure 1). As a cutting-edge biopharmaceutical technology, there are multiple gene therapies now FDA approved; with hundreds more in clinical trials, we’re likely to see many more of these therapies on the market soon.1 However, to keep up with the rapid pace of clinical research, developers are working to streamline the manufacturing and quality control process to improve quality and lower the cost of bringing these important drugs to market.
Developers use a multitude of analytical tests to develop gene therapies and optimize their manufacturing process. When developers get aberrant test results, they must be able to interpret where the problem lies. Did the manufacturing process produce an undesirable product, or is the analytical testing method unreliable? Analytical testing companies that have the infrastructure, personnel, and experience often partner with developers to tighten up analytical variability so that results of tests clearly indicate where there are opportunities to increase efficiency and product quality.1590077756898.jpg)
Figure 1. Gene delivery by recombinant viral vector. During gene therapy, viral capsids containing the therapeutic gene are taken up by the patient’s cells and the genetic material is delivered to the nucleus. There, the gene gets expressed as a protein necessary for the patient’s health. Credit: Avomeen.
Capsid complications
A major manufacturing challenge that developers contend with are impurities derived from the AAV vector itself (Figure 2). For example, empty AAV capsids represent a critical impurity that must be minimized during purification of the gene therapy product. These complete viral capsids do not contain DNA. When they are administered to a patient, they have the potential to cause life-threatening adverse events, such as immunotoxicity, without any potential therapeutic benefit. The same holds true for capsids that contain a partial copy of the therapeutic gene, which can be caused by incomplete viral packaging during manufacturing. Furthermore, empty capsids or capsids with partial packaging can compete with full capsids to infect the patient’s cells. The greater the quantity of inactive capsid in a dose, the higher the overall dose a patient will need for therapeutic effect. The higher the dose, the higher the risk of serious adverse events.
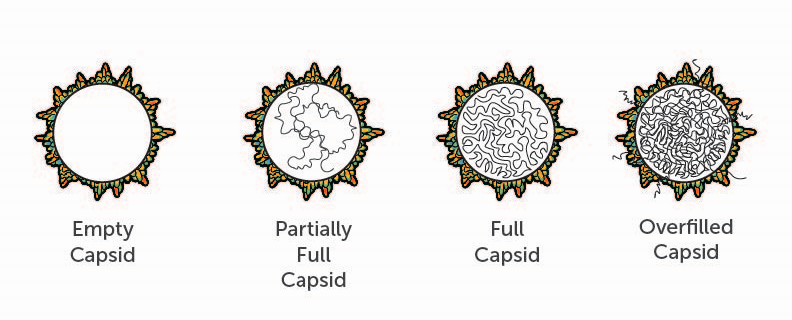
How do these impurities arise? Each recombinant AAV capsid is meant to deliver single-strand DNA encoding the therapeutic gene. AAV particles are assembled from 60 capsid proteins, with a defined stoichiometry and shape.2 However, there can be a high degree of fluctuation in the ratios of the three AAV capsid building blocks, with variable post-translational modifications. This complexity increases the likelihood of forming defective viral particles, incapable of delivering the desired gene therapy. At a molecular level, these impurities closely resemble the desired, active product. Developers must either minimize the generation of these and other undesirable products in the bioprocess during upstream manufacturing, and/or reduce or remove them from the final AAV product during downstream processes.
There are several ways to measure the empty/full capsid ratio, and as developers are establishing their chemistry, manufacturing and control (CMC) protocol, it is important that they choose an optimized method, as they must use that method for effective quality control from early process development to lot release and stability.3 Gene therapy developers may choose analytical ultracentrifugation to evaluate capsids, but while highly effective, this method is not as quantitative, robust or efficient as some newer methods. High-performance liquid chromatography (HPLC) using AAV full/empty analytical columns have been demonstrated to be highly effective at separating full, empty, and improperly filled capsids for robust quantification. Additionally, this method is higher throughput than ultracentrifugation, and requires less precious AAV sample to run.
Determining the product’s molecular fingerprint
Viral capsid proteins need to be made from the correct protein sequence, and they often require specific post-translational groups to be fully active in cells. These qualities of a gene therapy, and associated impurities, are characterized during process development and quality control through the process of peptide mapping.
Peptide mapping is accomplished through liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS). It maps out the primary protein sequence and post-translational modifications necessary for the AAV particle’s functionality in cells. The map can then be used for identity testing to monitor any changes in capsid production or impurity levels in relation to changes in manufacturing strategy. That way, while optimizing procedures and scaling up production, developers can ensure that the AAV will be consistently effective when administered to patients.
Evaluating potency and impurities
Finally, a gene therapy must be analyzed for its ability to transduce cells and tested for cytotoxic impurities. These tests give a functional readout of the gene therapy, revealing if the therapeutic gene delivered would be functional in the context of living cells.
Cellular potency is evaluated by transducing cells with the AAV product and then measuring a “phenotypic” or functional outcome due to the transduction. Developing these tests can be challenging because there is no one-size-fits-all test that will give developers the answers they need. Developers often draw on the experience of analytical labs to determine how to best evaluate their AAV product’s transduction efficiency.
A gene therapy in development must also be tested to ensure that it is free of residual, process-related impurities such as polyethylenimine, iodixanol, poloxamer, and other excipients that must be removed in the final product to ensure safety. Few research and manufacturing facilities have the equipment and expertise necessary to perform this kind of testing, and it is advisable to find one that has experience testing polymers, extractables and leachables to examine if components of the manufacturing equipment or drug’s packaging are not contaminating the final product.
Cutting costs to increase accessibility
Gene therapies are notoriously expensive compared to other drugs.4 However, the cost of a one-time cure is more comparable to that of a lifetime of treatment than one might expect.5 Developers make a large, upfront investment in drug research, development and manufacturing that trickles down to patients, but labs that specialize in analytical testing aim to streamline the process – reducing the burden for everyone.
As gene therapy is an emerging field, there is an unmet need for guidelines to improve efficiency of gene therapy CMC testing. However, headway is being made to standardize processes for better comparison of strength, potency, and particle-to-infectivity ratio of vectors across labs and studies, to ultimately establish a common dosage unit. While two AAV serotype reference standards are available to support this effort, there is a call to develop more robust analytical assays that will ensure consistency for quality control during gene therapy development and subsequent release to patients. 6
Furthermore, because of the newness of the technology, industry standards for analytically evaluating the quality of gene therapeutics are constantly changing. As recently as January 2020, the FDA released seven new guidances for developers.7 Remarkably, these are seen as a kind of minimum requirement by early gene therapy players, who, as they develop new therapies, are pushing themselves to reach an even higher standard of quality and share key findings to shape future industry standards.
As fast-paced as the gene therapy field is now, it stands to become a true race to the finish line to bring new gene therapies to market in the near future. Regulatory bodies are becoming more familiar with reviewing gene therapies, and the road to commercialization will move more quickly. There is no denying that gene therapies will bring incredible benefits to patients, but it will be crucial to improve manufacturing efficiency and lower costs to make gene therapies more accessible to the patients who need them.
References
1. Colasante, W., Diesel, P., and Gerlovin, Lev. (2018). New Approaches To Market Access And Reimbursement For Gene And Cell Therapies. Cell & Gene. Retrieved from: https://www.cellandgene.com/doc/new-approaches-to-market-access-and-reimbursement-for-gene-and-cell-therapies-0001
2. Fraser Wright, J. (2014). Product-Related Impurities in Clinical-Grade Recombinant AAV Vectors: Characterization and Risk Assessment. Biomedicines, 2, 80-97; doi:10.3390/biomedicines2010080
3. U.S. Food & Drug Administration (2019). Guidance for Human Somatic Cell Therapy and Gene Therapy. Retrieved from: https://www.fda.gov/animal-veterinary/guidance-industry/chemistry-manufacturing-and-controls-cmc-guidances-industry-gfis
4. Stein, R. (2019). At $2.1 Million, New Gene Therapy Is The Most Expensive Drug Ever. NPR. Retrieved from: https://www.npr.org/sections/health-shots/2019/05/24/725404168/at-2-125-million-new-gene-therapy-is-the-most-expensive-drug-ever
5. Cohen, J.T, Chambers, J. D., Silver, M. C., Lin, P., Neumann, P.J. (2019). Putting The Costs And Benefits Of New Gene Therapies Into Perspective. Health Affairs. Retrieved from: https://www.healthaffairs.org/do/10.1377/hblog20190827.553404/full/
6. ATCC (accessed May, 2020) ATCC Virus Reference Materials. Retrieved from: https://www.atcc.org/en/Standards/Standards_Programs/ATCC_Virus_Reference_Materials.aspx#
7. U.S. FDA (2020). FDA Details Policies on Gene Therapies in Seven Guidances. Retrieved from: https://www.fdanews.com/articles/195767-fda-details-policies-on-gene-therapies-in-seven-guidances

