Cryo-EM and CX-MS: The Power Couple of Proteomics


Complete the form below to unlock access to ALL audio articles.
An ambitious new workflow has married two cutting-edge proteomic analysis techniques to improve imaging of large protein assemblies. The combination of electron cryo-microscopy (cryo-EM) and cross-linking coupled mass spectrometry (CX-MS) allows deeper interrogation of protein structures to a level of detail that the techniques are incapable of obtaining on their own.
The new technical combination has been reviewed in Current Opinion in Structural Biology, authored by Martin Luther University of Halle-Wittenberg’s Carla Schmidt and the Max Planck Institute for Biophysical Chemistry’s Henning Urlaub.
CX-MS and Cryo-EM: Techniques on the Rise
Mapping the structure of a protein is a seriously complicated process. In addition to the covalent peptide bonds that link amino acids together in peptide chains, non-covalent bonds, such as van-der-Waals interactions and electrostatic interactions, form protein-protein interactions both within and between proteins. Currently, there is no easy way of identifying where these interactions are in a protein’s structure. CX-MS cross-links regions in a protein that are non-covalently bound, allowing visualization of these otherwise hidden connections.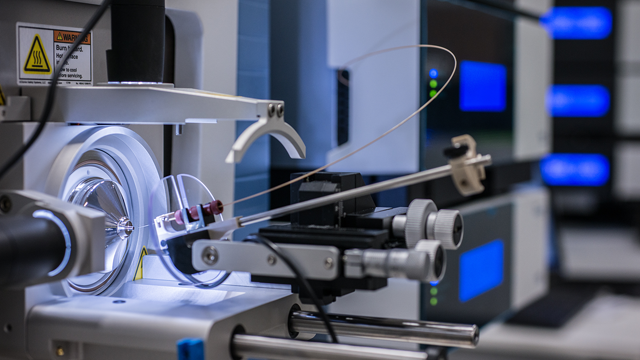 State-of-the-art Fourier-transform (FT) electrospray-ionisation mass spectrometer for the quantitative analysis of molecules. Tiny quantities of proteins and their fragments are separated by liquid chromatography and sprayed directly into the mass spectrometer. The screen shows the position of the spray nozzle (capillary needle made of silica glass) from which the molecules emerge in front of the opening of the mass spectrometer. Credit: Irene Böttcher-Gajewski / Max Planck Institute for Biophysical Chemistry.
State-of-the-art Fourier-transform (FT) electrospray-ionisation mass spectrometer for the quantitative analysis of molecules. Tiny quantities of proteins and their fragments are separated by liquid chromatography and sprayed directly into the mass spectrometer. The screen shows the position of the spray nozzle (capillary needle made of silica glass) from which the molecules emerge in front of the opening of the mass spectrometer. Credit: Irene Böttcher-Gajewski / Max Planck Institute for Biophysical Chemistry.
To link these protein regions, chemical cross-linkers are used. Only recently have cross-linkers been created that can be cleaved in a mass spectrometer, which has opened up CX-MS’s potential to be used combination with cryo-EM. These cross-linkers include disuccinimidyl suberate (DSS) and bis(sulfosuccinimidyl) suberate (BS3) which target the amine group of lysine residues in the peptide chain. Further innovation has yielded crosslinkers such as 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methyl-morpholinium chloride (DMTMM), which directly links the carboxyl group in aspartate and glutamate with primary amines.
Cryo-EM has its roots in transmission electron microscopy (TEM). Like TEM, it reconstructs a 3D image of a samples from 2D image slices. To protect the samples from radiation damage (a major concern for biological samples such as proteins) and the vacuum created within the electron microscope, samples are studied at low temperatures in a state of vitrification (essentially turning the sample into a non-crystallized glass) which avoids the production of damaging ice crystals. Recent advances have improved the resolution of Cryo-EM, meaning that resolution of the latest published 3D-EM images sits at 3-5Å.
Coupling Cryo-EM and Crosslinking
After CX-MS has identified cross-linked peptides, data from cryo-EM assays can be integrated with the CX-MS data. CX-MS can assist in reconstruction process of stitching together a 3D image of a protein from 2D image slices, by providing constraints which limit the maximum possible distance between amino-acids in a peptide, helping identify and validate their location on the reconstructed protein complex.Further innovation has sought to automate the process of fitting cross-linked proteins onto a 3D-EM map, and to help reconcile the defined structure of a cross-linked protein with the actual structure of a native protein, which is constantly in a state of flux. This flux, or conformational heterogeneity, enables proteins to bind to many partners depending on what role they are required to fill.
Applications of Cryo-EM/CX-MS
The cryo-EM/CX-MS workflow was used to image the spliceosome, a protein complex present in eukaryotic cells which chops and processes a pre-mRNA to produce a mature mRNA molecule free of introns. The spliceosome structure is constantly changing during the splicing process, and cryo-EM/CX-MS has proved vital to visualizing the sub-complexes of the spliceosome. The technique combination has also been used to identify membrane-embedded proteins, with a cryo-EM identification step followed up by CX-MS location validation.Whilst the techniques do not always need to be combined – for assemblies with a high cryo-EM resolution of below 3.5Å, CX-MS is unnecessary as the resolution is high enough to easily model the entire protein structure – a cryo-EM/CX-MS workflow has found a useful and substantial niche in structural analysis. Improvements still need to be made in the cross-linkers used, to penetrate to regions deep in proteins’ interiors, and to overcome a reliance on reactive residues to form crosslinks, but cryo-EM/CX-MS is a combination that has potential to change entirely how the structural biology field approaches protein complex analysis.

