Proteomics: Principles, Techniques and Applications
Unlike the genome, the composition of the proteome is in a constant state of flux over time and throughout the organism.


Complete the form below to unlock access to ALL audio articles.
Contents
What are the key questions that proteomics can answer?
- Low-throughput methods:
- 1. Antibody-based methods
- 2. Gel-based methods
- 3. Chromatography-based methods
- High-throughput methods:
- 1. Analytical, functional and reverse-phase microarrays
- 2. Mass spectrometry-based proteomics
What is the proteome?
Proteins are biological molecules made up of building blocks called amino acids. Proteins are essential to life, with structural, metabolic, transport, immune, signaling and regulatory functions among many other roles.1
The term “proteome” was coined by an Australian PhD student, Marc Wilkins, in a 1994 symposium held in Siena, Italy.2 It is a blanket term that refers to all of the proteins that an organism can express. Each species has its own, unique proteome.
Unlike the genome (the complete set of genes within each organism), the composition of the proteome is in a constant state of flux over time and throughout the organism.3 Therefore, when scientists refer to the proteome, they are also sometimes referring to the proteome at a given point in time (such as the embryo versus the mature organism), or to the proteome of a particular cell type or tissue within the organism.
 Figure 1: There are approximately 20,000 genes in the human genome, ~100,000 transcripts in the human transcriptome and over >1000000 proteoforms in the human proteome. Credit: Technology Networks
Figure 1: There are approximately 20,000 genes in the human genome, ~100,000 transcripts in the human transcriptome and over >1000000 proteoforms in the human proteome. Credit: Technology Networks
What is proteomics?
Proteomics is the study of the proteome – investigating how different proteins interact with each other and the roles they play within the organism.4
Although protein expression can be inferred by studying the expression of mRNA, which is the middle man between genes and proteins, mRNA expression levels do not always correlate well with protein expression levels.1,3 Furthermore, the study of mRNA does not consider protein posttranslational modifications, cleavage, complex formation and localization, or the many variant mRNA transcripts that can be produced; all of which are key to protein function.

The first experiments that fit the label of “proteomic” studies were performed in 1975 with the development of 2D protein electrophoresis.5
However, truly high-throughput identification of multiple proteins per sample only became possible with the development of mass spectrometry (MS) technology over 20 years later.6
Since then, the sensitivity and accuracy of MS have advanced to the point where proteins can be reliably detected down to the attomolar range (1 target protein molecule per 1018 molecules),7 and various other proteomic techniques have been developed and optimized.
What are the key questions that proteomics can answer?
Broadly speaking, proteomic research provides a global view of the processes underlying healthy and diseased cellular processes at the protein level.3,4 To do this, each proteomic study typically focuses on one or more of the following aspects of a target organism’s proteome at a time to slowly build on existing knowledge:
Protein identification | Which proteins are normally expressed in a particular cell type, tissue or organism as a whole, or which proteins are differentially expressed? |
Protein quantification | Measures total (“steady-state”) protein abundance, as well as investigating the rate of protein turnover (i.e., how quickly proteins cycle between being produced and undergoing degradation). |
Protein localization | Where a protein is expressed and/or accumulates is just as crucial to protein function as the timing of expression, as cellular localization controls which molecular interaction partners and targets are available. |
Post-translational modifications | Post-translational modifications can affect protein activation, localization, stability, interactions and signal transduction among other protein characteristics, thereby adding a significant layer of biological complexity. |
Functional proteomics | This area of proteomics is focused on identifying the biological functions of specific individual proteins, classes of proteins (e.g., kinases) or whole protein interaction networks. |
Structural proteomics | Structural studies yield important insights into protein function, the “druggability” of protein targets for drug discovery, and drug design. |
Protein-protein interactions | Investigates how proteins interact with each other, which proteins interact, and when and where they interact. |
Proteomics: Techniques
Low-throughput methods:
1. Antibody-based methods
Techniques such as ELISA (enzyme-linked immunosorbent assay) and western blotting rely on the availability of antibodies targeted toward specific proteins or epitopes to identify proteins and quantify their expression levels.
2. Gel-based methods
Two-dimensional gel electrophoresis (2DE or 2D-PAGE), the first proteomic technique developed, uses an electric current to separate proteins in a gel based on their charge (1st dimension) and mass (2nd dimension). Differential gel electrophoresis (DIGE) is a modified form of 2DE that uses different fluorescent dyes to allow the simultaneous comparison of two to three protein samples on the same gel. These gel-based methods are used to separate proteins before further analysis by e.g., mass spectrometry (MS), as well as for relative expression profiling.
3. Chromatography-based methods
Chromatography-based methods can be used to separate and purify proteins from complex biological mixtures such as cell lysates. For example, ion-exchange chromatography separates proteins based on charge, size exclusion chromatography separates proteins based on their molecular size, and affinity chromatography employs reversible interactions between specific affinity ligands and their target proteins (e.g., the use of lectins for purifying IgM and IgA molecules). These methods can be used to purify and identify proteins of interest, as well as to prepare proteins for further analysis by e.g., downstream MS. 8
High-throughput methods:
1. Analytical, functional and reverse-phase microarrays
Protein microarrays apply small amounts of sample to a “chip” for analysis (this is sometimes in the form of a glass slide with a chemically modified surface). Specific antibodies can be immobilized to the chip surface and used to capture target proteins in a complex sample. This is termed an analytical protein microarray, and these types of microarray are used to measure the expression levels and binding affinities of proteins in a sample. Functional protein microarrays are used to characterize protein functions such as protein–RNA interactions and enzyme-substrate turnover. In a reverse-phase protein microarray, proteins from e.g., healthy vs. diseased tissues or untreated vs. treated cells are bound to the chip, and the chip is then probed with antibodies against the target proteins.
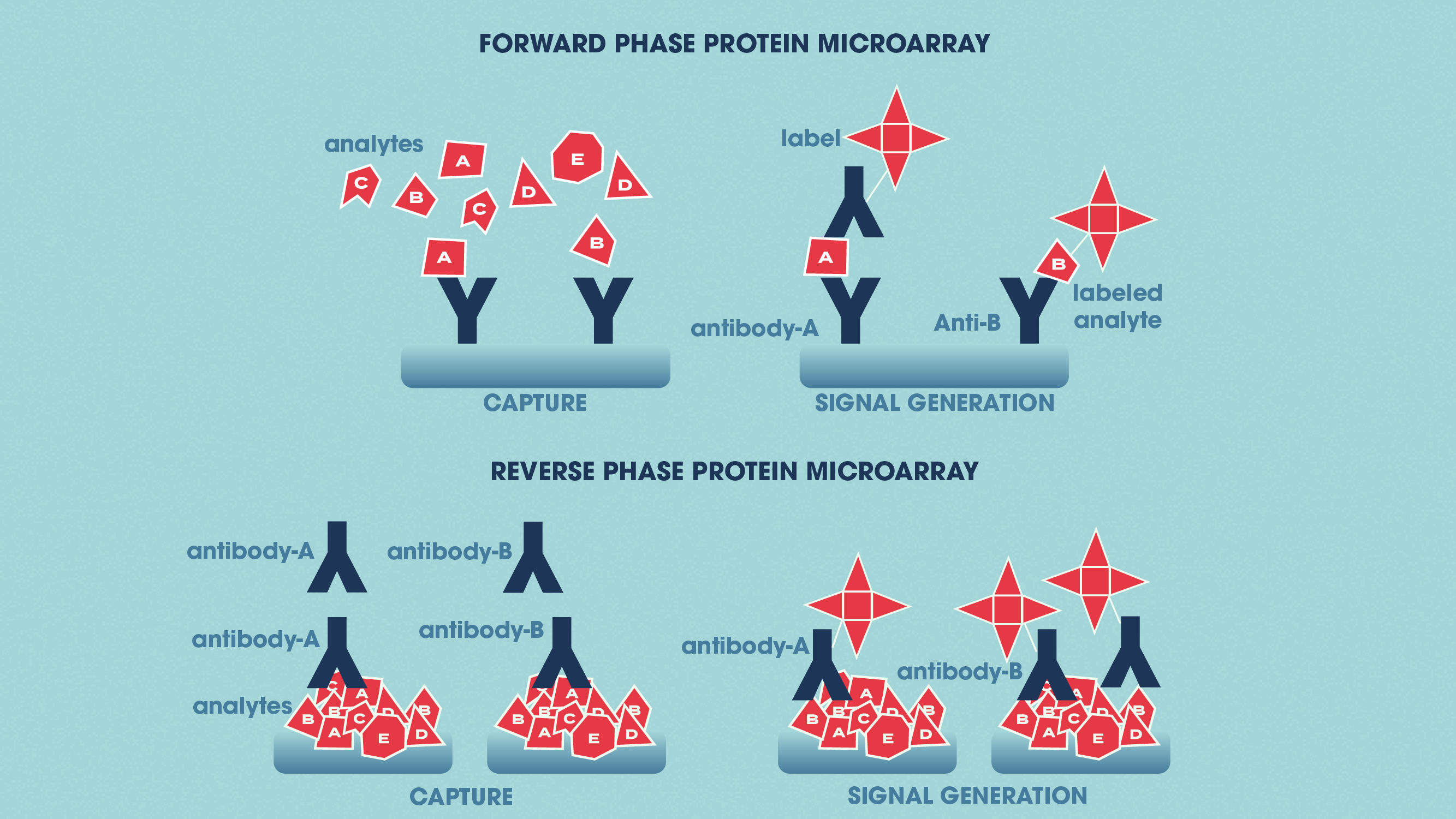 Figure 2: The differences between forward phase and reverse phase protein microarrays. Credit: Technology Networks.
Figure 2: The differences between forward phase and reverse phase protein microarrays. Credit: Technology Networks.
2. Mass spectrometry-based proteomics
There are several “gel-free” methods for separating proteins, including isotope-coded affinity tag (ICAT), stable isotope labeling with amino acids in cell culture (SILAC) and isobaric tags for relative and absolute quantitation (iTRAQ). These approaches allow for both quantitation and comparative/differential proteomics. There are also other, less quantitative techniques such as multidimensional protein identification technology (MudPIT), which offer the advantages of being faster and simpler. Other gel-free, chromatographic techniques for protein separation include gas chromatography (GC) and liquid chromatography (LC). 8,9
Mass spectrometry workflow
Regardless of how the protein sample is separated, the downstream MS workflow comprises three main steps:
1. The proteins/peptides are ionized by the ion source of the mass spectrometer.
2. The resulting ions are separated according to their mass to charge ratio by the mass analyze.
3. The ions are detected.
When using gel-free techniques upstream of MS such as iTRAQ or SILAC, the samples are used directly for input into the mass spectrometer. When using gel-based techniques, the protein spots are first cut out of the gel and digested before being either separated by LC or directly analyzed by MS.
There are two main ionization sources, namely:
- Matrix-assisted laser desorption/ionization (MALDI)
- Electrospray ionization (ESI)
 and Electrospray ionization (ESI).") Figure 3: The two main ionization sources in MS-based proteomics. Credit: Technology Networks.
Figure 3: The two main ionization sources in MS-based proteomics. Credit: Technology Networks.
Other, less common sources include chemical ionization, electron impact and glow discharge ionization.
There are four main mass analyzers:
- Time-of-flight (TOF)
- Ion trap
- QuadrupoleFourier-transform ion cyclotron (FTIC)
- Electrostatic sector and magnetic sector are two other, less commonly adopted mass analyzers.
What is tandem MS?
Peptides can be subjected to multiple rounds of fragmentation and mass analysis – a process termed tandem-MS, MS/MS or MSn. By combining the same or different mass analyzers in tandem, such as quadrupole-TOF (Q-TOF) or triple-quadrupole (QQQ) MS, the strengths of different mass analyzers can be leveraged to further improve the capacity for proteome-wide analysis. Simple MS setups such as MALDI-TOF are only used for peptide mass measurements, whereas tandem mass spectrometers are used to determine peptide sequences.
Top-down proteomics vs. bottom-up proteomics
In top-down proteomics, the proteins in a sample of interest are first separated before being individually characterized.1,10
With bottom-up proteomics – also termed “shotgun” proteomics – all the proteins in the sample are first digested into a complex mixture of peptides, and these peptides are then analyzed to identify which proteins were present in the sample.1,10
Approach | Description | Method |
Top-down proteomics | Proteins in a sample of interest are first separated before being individually characterized. | Protein separation is performed based on mass and charge with e.g., 2DE, DIGE or MS. When using 2D electrophoresis techniques, the proteins are first resolved on the gel and then individually digested into peptides that are analyzed by a mass spectrometer. When using MS directly, the undigested sample containing the whole proteins is injected into the mass spectrometer, the proteins are separated, and individual proteins are then selected for digestion and a further round of MS for analysis of the peptides. |
Bottom-up proteomics, or "shotgun proteomics" | All the proteins in the sample are first digested into a complex mixture of peptides, and these peptides are then analyzed to identify which proteins were present in the sample. | Proteins are first digested, and the digested peptide mixture is fractionated and subjected to MS, frequently in an LC-MS/MS configuration. The resulting peptide sequences are compared to existing databases using automated search algorithms. These search engines match the experimentally obtained peptide spectra to the predicted spectra of proteins produced by in silico digestion (this is called “peptide-spectrum matching”). There are several different bottom-up workflows possible, including data-dependent and data-independent methods, as well as hybrids of these. |
Both the top-down and bottom-up approaches have their own set of advantages and disadvantages and applications to which each is more suited.10,11 For example, top-down MS is more appropriate for research on different PTMs and protein isoforms. However, it is limited by difficulties inherent in separating complex mixtures of proteins and the decreasing sensitivity of MS toward larger proteins (particularly > 50 to 70 kDa).1
In contrast, while the peptides used in bottom-up MS (~5 to 20 amino acids in length) are much easier to fractionate, ionize and fragment, this approach provides an indirect measure of the proteins originally present in samples and relies heavily on inference.1 A hybrid “middle-down” approach has been developed, which employs larger peptide fragments than conventional bottom-up proteomics, thereby potentially allowing more unique peptide matches.

Figure 4: The differences between top-down and bottom-up proteomics. Credit: Technology Networks.
Data analysis in proteomics
Proteomic studies, particularly those employing high-throughput technologies, can generate huge amounts of data.12 In addition to the sheer quantity of data produced, proteomic data analysis can also be relatively complex for certain techniques such as shotgun MS.13 Adding to this complexity is the range of bioinformatics tools available for proteomic analyses.14-17

Proteomic researchers are faced with many hurdles when attempting to optimize how they warehouse and analyze their proteomic data.12
When planning proteomic experiments, scientists need to factor in not only the costs of the reagents and laboratory equipment but also that of data storage and analysis, and they have to appraise the level of bioinformatics skills and computational resources required.
Proteomic studies often require multiple data processing and analysis steps that need to be performed in a specific sequence.12 To address this need, researchers are increasingly assembling the needed scripts, tools and software into customized proteomic analysis pipelines suited to their particular research questions.
Applications of proteomics
The applications of proteomics are incredibly numerous and varied. The table below lists just some of these applications and provides links to examples of studies using these approaches.
Proteomics application | Description and examples |
Personalized medicine | Tailoring disease treatment to each patient based on their genetic and epigenetic makeup, so as to improve efficacy and reduce adverse effects. While genomics and transcriptomics have been the main focus of such studies to date, proteomics data will likely add a further dimension for patient-specific management. |
Biomarker discovery | Identification of protein markers e.g., for the diagnosis and prognosis of glioblastoma, and evaluating patients’ response to therapeutic interventions such as stem cell transplantation. |
Drug discovery and development | Identifying potential drug targets, examining the druggability of selected protein targets, and developing drugs aimed at candidate therapeutic protein targets (e.g., for hepatocellular carcinoma). |
Systems biology | System-wide investigations of disease pathways and host–pathogen interactions to identify potential biomarkers and therapeutic targets; system-wide investigations of drug action, toxicity, resistance and efficacy. |
Agriculture | Investigations of plant–pathogen interactions, crop engineering for increased resilience to e.g., flooding, drought and other environmental stresses. |
Food science | Food safety and quality control, allergen detection and improving the nutritional value of foods. |
Paleoproteomics | The study of ancient proteins to further our understanding of evolution and archeology. |
Astrobiology | Investigations of how mammals’ immune systems may respond to exo-microbes found in space and studies of the prebiotic organic matter found on meteorites. |
The future of proteomics
Currently, proteomic workflows rely heavily on MS.1 As powerful as this technology has proven, researchers are now looking ahead to a future for proteomics that lies “beyond MS.” Despite the attomolar sensitivity of MS, millions of the target molecule still need to be present in the sample for it to be detected. This implies that low-concentration target molecules (e.g., serum biomarkers) can be undetectable in complex milieu such as human serum unless first enriched for.
Scientists are still searching for the holy grail of high-throughput proteomic techniques that 1) has excellent sensitivity across the dynamic range of the target proteome (e.g., 107 for the human proteome), 2) can directly read entire protein sequences and identify their PTMs, and, therefore, 3) does not need to draw inferences from databases of theoretical protein matches.1
There are several promising technologies that, while currently hampered by limitations in sensitivity, throughput or cost, may yet come to dominate the proteomic field.1 These include nascent fluorescent fingerprinting methods and yet-to-be-developed subnanopore arrays for the high-throughput single-molecule sequencing of proteins.
Along with the expected advances in proteomic techniques, approaches to proteomic data analysis are expected to evolve just as rapidly. For example, there is a strong impetus towards developing data technologies such as cloud computing, software containers and workflow systems, which will “democratize” access to top-notch computing resources for proteomic data analysis regardless of researchers’ location, IT infrastructure or computational expertise.12,18,19

