Recent Advances in Single-Cell Genomics Techniques


Complete the form below to unlock access to ALL audio articles.
Single-cell genomics and its contrast with bulk tissue genomics analyses
After the advent of next-generation sequencing (NGS), high-throughput measurements of biomolecules in tissues, especially RNA and DNA, developed rapidly. These tissue-level genomics approaches, often referred to as bulk genomics techniques, enabled a revolution in the approach to studying normal biological processes and diseases.
However, bulk genomics methods have a major limitation: the amount of biomolecules they require as an input restrict the analysis to RNA or DNA isolated from populations of cells in a tissue. This leads to a loss of cellular resolution when performing bulk tissue genomics measurements; a major drawback considering that cells are functional units of tissues and organs, and different cell types express a highly specific signature of RNA, epigenetic marks and proteins.Single-cell genomics methods, such as single cell RNA sequencing (scRNA-seq), involve labeling biomolecules originating from individual cells (Figure 1), therefore enabling high-throughput molecular analysis at single-cell resolution.
 Figure 1. Bulk RNA sequencing vs Single-cell RNA sequencing. Image Credit: Dmitry Velmeshev.
Figure 1. Bulk RNA sequencing vs Single-cell RNA sequencing. Image Credit: Dmitry Velmeshev.
Differences between bulk and scRNA-seq
Bulk RNA sequencing (top, Figure 1) measures tissue gene expression levels that are average expression profiles of individual cells. By utilizing this approach to compare expression levels for a hypothetical gene "X" between two conditions, one might conclude there is no significant difference. scRNA-seq (bottom) preserves the information about the cell of origin for each RNA molecule measured. Using scRNA-seq to measure the level of gene "X" in the same tissue and conditions, one might conclude that expression of gene "X" is perturbed in a specific cell type, a phenomenon that would be masked in the bulk measurement.
Molecular readouts and applications of single-cell genomics
scRNA-seq is the most widely used single-cell genomics method that has been developing especially rapidly.
By measuring RNA molecules from thousands of individual cells1, scRNA-seq permits identification of known and novel cell types and the investigation of cell state transitions during normal organ development and disease.
“Single-cell RNA sequencing is our first-choice technique when it comes to identifying transcriptomic profiles of rare cell types for which no reliable markers exist”, comments Wei Huang, a postdoctoral scholar at the University of California, San Francisco, who studies glia cells types in the developing human brain. “Using scRNA-seq does not require purifying a priori known cell populations since they can be discovered in an unbiased way based on single-cell gene expression profiles.”
However, the arsenal of single-cell genomics is not limited to profiling RNA. More recently, efforts to measure epigenetic signatures in single-cells led to the development of a number of novel techniques, such as single-cell assay for transposase-accessible chromatin using sequencing (scATAC-seq).
scATAC-seq profiles regions of DNA in single cells that are in so-called "open states" - indicative of these regions being epigenetically activated. Recently, scATAC-seq has been combined with droplet-based sequencing, allowed profiling of open chromatin regions in thousands of individual cells2. In addition to analyzing open-chromatin regions, other efforts are directed at investigating DNA methylation3 and histone modification4 in single cells.
Combination of scRNA-seq Strategies to Untangle Complex Cell Populations
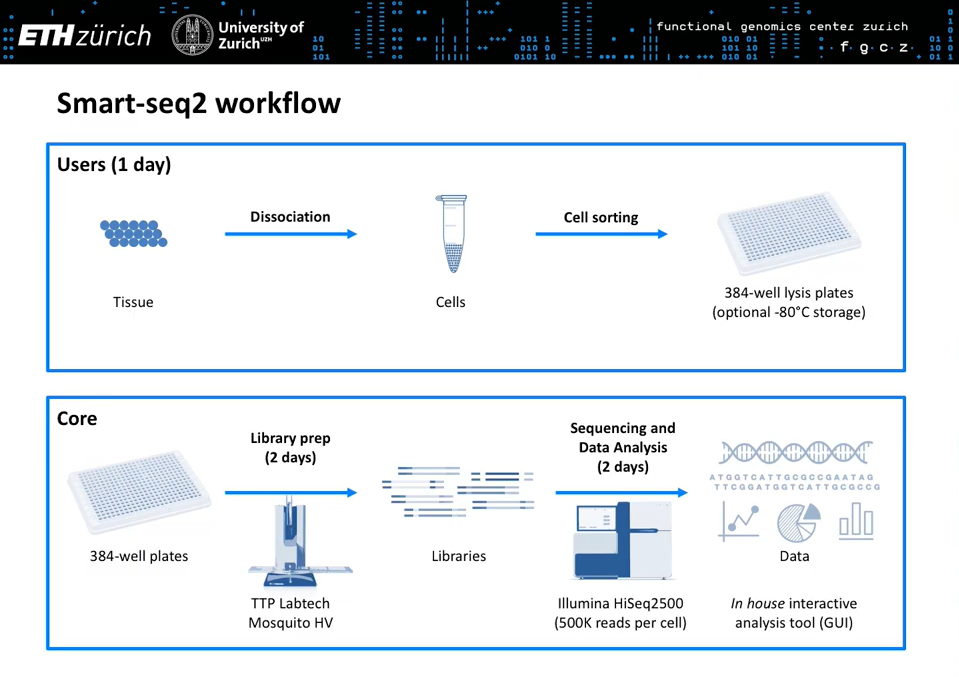
This workshop held at the EMBO Single Cell Biology 2019 conference showcases the technologies and strategies for single cell RNA-seq that scientists at the Functional Genomics Center Zurich (FGCZ) use to answer a variety of challenging biological questions.
Watch VideoSponsored Content
Single-cell genomics to study cell types of mammalian organ systems
In the last five years, scRNA-seq has been utilized to uncover the cellular and molecular composition of a range of mammalian organ systems.
Studies of the central nervous system (CNS) especially benefited from single-cell genomics approaches due to the variety of cell types in the CNS, for many of which no reliable markers are currently available. These studies include analyses of the developing human cerebral cortex5, which revealed the signatures of cardinal cell types and molecular dynamics during neocortex development, and of the entire CNS of the mouse6, which produced a comprehensive atlas of neuronal and glial cell types in the mouse.
Other studies revealed the diversity of cell types in the human liver7, the immune system8 and the mouse small intestine9, all highlighting the variety of resident immune cells in various organ systems.
Studies of cellular heterogeneity in the mammalian brain have not been limited to scRNA-seq and include scATAC-seq analysis of the developing mouse forebrain10 to identify genomic regulatory elements that are guiding brain development, as well as single-cell DNA sequencing11 of the human brain tissue, which suggested that somatic mutations in single neurons can be utilized to trace their developmental lineage and predict susceptibility to disease.
“The human brain contains a huge variety of cell types, for many of which we lack reliable markers that can be used to isolate them” says Huang. “Therefore, the human brain is the organ for which applications of single-cell genomics have been especially instrumental.”
Novel insights into the mechanisms of human diseases with single-cell genomics.
Single-cell genomics not only allow for insight into the heterogeneity of tissues and organs in normal development and function, but also enable the study of disease processes in specific cell types.
One of the first human diseases to be approached using single-cell genomics methods was glioblastoma multiform (GBM). scRNA-seq analysis of GBM revealed molecular and cellular heterogeneity of this cancer, including cell populations expressing markers of stemness and thus potentially representing cancer stem cells.12
More recently, the advent of single-nucleus RNA sequencing13 (snRNA-seq) allowed the extension of single-cell transcriptomics analyses to human diseases for which live tissue is not obtainable. One such study conducted single-cell analysis of molecular pathology in the brain of patients with autism spectrum disorder (ASD), which demonstrated that specific types of neurons in the cerebral cortex could be more impacted by the changes associated with ASD.14
Another project focused on a different disease of the human brain, multiple sclerosis (MS). Applying snRNA-seq to post-mortem brain tissue samples of patients with MS and healthy controls, a team of scientists from the University of California, San Francisco, were able to identify gene expression changes in specific cell types that are associated with progression of MS.15
“Thanks to this innovative technique, we were able for the first time to get insight into how different cell types in the cerebral cortex respond to MS lesions”, says Lucas Schirmer, Ph.D, one of the two lead authors on the study. “The results of this study represent a wealth of potential drug targets, biomarkers and key mechanistic players in MS pathology, on which we are following up in my lab”, adds Dr. Schirmer.
Combinatorial molecular profiling in single cells
One of the latest trends in single-cell genomics is obtaining a combination of multiple molecular readouts from thousands of single cells. As such, the application of single-cell combinatorial indexing (sci) has allowed for simultaneous analysis of the open chromatin and RNA profiles of individual cells.
This technology, coined sci-CAR (single-cell combinatorial indexing for chromatin accessibility and mRNA analysis), allows researchers to link the transcriptomic and open-chromatin profiles in specific cell types of the mouse kidney and identify cell type-specific open-chromatin regions.16
Another approach combined profiling of RNA and protein epitopes in single cells and was published in two17 back-to-back18 articles. These methods enable another level of insight in addition to gene expression profiling, allowing analysis of protein levels in specific cell types in an unbiased way, which provides a more direct indication of cellular functions.
The area of single-cell genomics is developing at an unprecedented speed, with new insights into mechanisms of normal organismal functions and disease already gained thanks to these new technologies. Undouble, we are observing a revolution in the way basic biology and translational questions are approached, and more exciting methods and discoveries will emerge in the near future.
References
1. Macosko, E. Z. et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161, 1202-1214, doi:10.1016/j.cell.2015.05.002 (2015).
2. Lareau, C. A. et al. Droplet-based combinatorial indexing for massive-scale single-cell chromatin accessibility. Nat Biotechnol 37, 916-924, doi:10.1038/s41587-019-0147-6 (2019).
3. Karemaker, I. D. & Vermeulen, M. Single-Cell DNA Methylation Profiling: Technologies and Biological Applications. Trends Biotechnol 36, 952-965, doi:10.1016/j.tibtech.2018.04.002 (2018).
4. Ku, W. L. et al. Single-cell chromatin immunocleavage sequencing (scChIC-seq) to profile histone modification. Nat Methods 16, 323-325, doi:10.1038/s41592-019-0361-7 (2019).
5. Nowakowski, T. J. et al. Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science 358, 1318-1323, doi:10.1126/science.aap8809 (2017).
6. Zeisel, A. et al. Molecular Architecture of the Mouse Nervous System. Cell 174, 999-1014.e1022, doi:10.1016/j.cell.2018.06.021 (2018).
7. MacParland, S. A. et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat Commun 9, 4383, doi:10.1038/s41467-018-06318-7 (2018).
8. Stubbington, M. J. T., Rozenblatt-Rosen, O., Regev, A. & Teichmann, S. A. Single-cell transcriptomics to explore the immune system in health and disease. Science 358, 58-63, doi:10.1126/science.aan6828 (2017).
9. Haber, A. L. et al. A single-cell survey of the small intestinal epithelium. Nature 551, 333-339, doi:10.1038/nature24489 (2017).
10. Preissl, S. et al. Single-nucleus analysis of accessible chromatin in developing mouse forebrain reveals cell-type-specific transcriptional regulation. Nat Neurosci 21, 432-439, doi:10.1038/s41593-018-0079-3 (2018).
11. Lodato, M. A. et al. Somatic mutation in single human neurons tracks developmental and transcriptional history. Science 350, 94-98, doi:10.1126/science.aab1785 (2015).
12. Patel, A. P. et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396-1401, doi:10.1126/science.1254257 (2014).
13. Lake, B. B. et al. Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science 352, 1586-1590, doi:10.1126/science.aaf1204 (2016).
14. Velmeshev, D. et al. Single-cell genomics identifies cell type-specific molecular changes in autism. Science 364, 685-689, doi:10.1126/science.aav8130 (2019).
15. Schirmer, L. et al. Neuronal vulnerability and multilineage diversity in multiple sclerosis. Nature, doi:10.1038/s41586-019-1404-z (2019).
16. Cao, J. et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science 361, 1380-1385, doi:10.1126/science.aau0730 (2018).
17. Stoeckius, M. et al. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods 14, 865-868, doi:10.1038/nmeth.4380 (2017).
18. Peterson, V. M. et al. Multiplexed quantification of proteins and transcripts in single cells. Nat Biotechnol 35, 936-939, doi:10.1038/nbt.3973 (2017).

