Accelerating Proteome Analysis With PASEF: A Unique and Sensitive Technique

Complete the form below to unlock access to ALL audio articles.
Understanding the proteome is a fundamental challenge of biology. Any large-scale, proteomics-related study will necessitate the ability to reliably identify as many components of the proteome as possible. High resolution mass spectrometry (MS)-based proteomics has developed into a highly effective tool for profiling thousands of proteins.
The sensitivity of mass spectrometers now on the market is enabling scientists to delve deeper into the entire proteome, to detect protein expression patterns and ultimately, to identify and characterize protein behavior under specific environmental, physiological or disease conditions. The applications of proteomics are therefore numerous, for example to identify clinical biomarkers in molecular diagnostics, to understand (antimicrobial) drug resistance, and for the large-scale analysis of post-translational modifications (PTMs).
A revolutionary new method, Parallel Accumulation Serial Fragmentation (PASEF), available on the timsTOF™ Pro mass spectrometer (Bruker Daltonics) is set to transform the field of proteomics by offering faster, more sensitive and more robust proteome analysis, giving scientists the potential to discover low-level, biologically significant proteins and validate them in research.
Obtaining insightful data
In contrast to traditional biochemical and biophysical methods, MS-based techniques can perform large-scale, systematic measurements of proteomes, to generate biological insights from the computational analysis of proteomics datasets [1]. Such methods enable the conclusive identification and accurate quantification of almost any protein that has been expressed.
There are two main methods for studying proteins by MS: in their intact form by top-down proteomics or using a bottom-up approach to digest the protein and identify the peptides. Top-down proteomics has the advantage of measuring any post-translational modifications (PTMs), enabling proteoforms to be fully characterized. However, carrying out high-throughput top-down proteomics is challenging for many laboratories, and it is more difficult to analyze proteins in comparison to peptides. Bottom-up proteomics involves the extraction of proteins from the source material and the digestion of these proteins into peptides, often with a sequence-specific enzyme such as trypsin, before separation using liquid chromatography (LC) and subsequent analysis with MS.
The primary method used in bottom-up proteomics is shotgun (or discovery) proteomics, using data-dependent acquisition (DDA) for unbiased and complete proteome coverage. It prioritizes the profiling of the most numerous peptides with a high degree of specificity, and the researcher does not need to know the identity of the expected proteins in advance. Mass spectrometers in DDA mode obtain mass spectra in a matter of seconds by scanning the entire mass range and by fragmenting as many peptides as possible within a specific time frame, which is ideal for discovery studies. However, a set-back of the DDA method is its sequencing speed and, therefore, only a percentage of eluting peptides can be fragmented. Improvements in MS technology and the introduction of trapped ion mobility spectrometry (TIMS) in combination with the PASEF method have significantly increased the sequencing speed of shotgun proteomics with DDA.
PASEF is an emerging method which enables several precursor ions to be stored in parallel in a trapped ion mobility device, which can then be followed with serial fragmentation. It allows hundreds of MS/MS events per second, at full sensitivity, providing a several-fold speed and sensitivity increase compared to traditional techniques [2], and increased DDA rates.
The higher sensitivity and higher speed shotgun proteomics enabled by PASEF technology make it ideally-suited for the analysis of samples where very limited sample is available, for example in biopsies. A completely open data file format allows researchers to work directly with the raw data when using software for shotgun proteomics analysis, such as MaxQuant. PEAKS Studio is another software package supporting MS data analysis, which has a novel de novo sequencing algorithm that is integrated into the traditional database searches to ensure a complete interpretation of raw spectral data. The solution handles the complexity of MS data, to advance proteomics research through peptide/protein identification and quantification, peptide mapping and PTM characterization (Figure 1).

Figure 1: Parallel Accumulation Serial Fragmentation (PASEF) for label-free quantitation (LFQ). Enhanced sensitivity, speed and reproducibility in ID rates are pre-requisites for enhanced LFQ performance. LFQ intensities of a Hela triplicate analysis (200 ng, 90 min gradient) with R2 > 0.99.
Simplifying complex signals
The PASEF technique can be exclusively applied on a TIMS quadrupole time-of-flight (QTOF) mass spectrometer, adding an additional dimension of separation to standard MS scans. Ion mobility spectrometry has been used in many applications – such as for the characterization of biomolecular structures and the high-throughput separation of complex mixtures – for many years, and these systems are often coupled to time-of-flight (TOF) MS. More recently, TIMS-QTOF MS has been developed for the separation and identification of complex protein digests.
For example, a group at the Max Planck Institute for Biochemistry has demonstrated the use of PASEF on TIMS-QTOF MS for shotgun proteomics. Figure 2 shows the result of timsTOF Pro analysis of a complete mixture of four digested proteins; alcohol dehydrogenase (ADH), bovine serum albumin (BSA), enolase, and phosphorylase b that were directly injected into the mass spectrometer [2]. Figure 2A right spectrum highlights the complexity of overlapping signals of the mass spectrum without ion mobility separation, which is simplified with TIMS separation (heatmap Figure 2A). The sequential isolation of four ions at different m/z that are separated by mobility after parallel accumulation is shown in Figure 2B, and by then applying PASEF, an MS/MS scan was performed on isolated precursors, leading to a typical ladder of fragmented ions (Figure 2C). Projected mass spectra are shown in the boxes on the right of the panels.
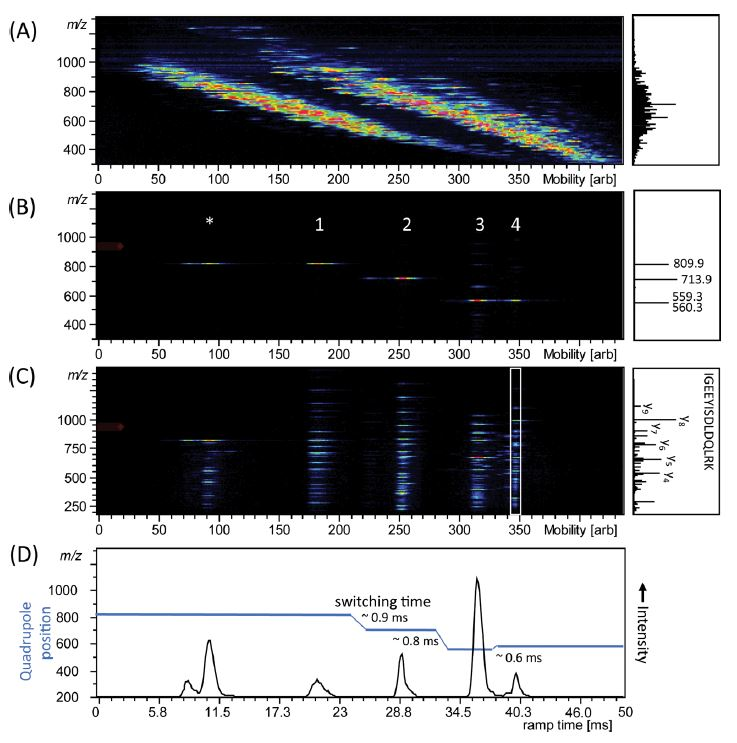
Figure 2: Trapped ion mobility spectrometry (TIMS)-quadrupole time-of-flight (QTOF) analysis of electrosprayed peptides from a combined tryptic digest of alcohol dehydrogenase (ADH), bovine serum albumin (BSA), phosphorylase b, and enolase. (A) Nested m/z and ion mobility distribution as detected in full scan MS. (B) Sequential isolation of four ions at different m/z that are separated by mobility after parallel accumulation. (C) Parallel accumulation−serial fragmentation (PASEF) of the precursors as isolated in (B). (D) Arrival time distribution of the summed fragment ions as observed by the PASEF method together with the quadrupole isolation mass as a function of the TIMS ramp time. Projected mass spectra are shown on the right of the panels, in (C) the fragments of the white box are projected.
The data from this study showed between a four- to ten-fold increase in MS/MS speed at full sensitivity using PASEF. But what is the impact of such an increase on typical shotgun proteomics data?
Clinical applications
In addition to its application in interaction proteomics and PTM-related proteomics, TIMS-QTOF MS is gaining momentum in clinical proteomics research. Protein mixtures derived from patient samples can be used to inform diagnosis and treatment of a number of diseases. Immunoassays are typically used for routine qualitative analysis, but this method cannot distinguish protein isoforms – a challenge addressed by MS-based proteomics. TIMS-QTOF MS with the PASEF method addresses other challenges faced by bottom-up proteomics for example, clinical samples require reproducible, robust and high-throughput sample preparation workflows. The high specificity, sensitivity and throughput provided by TIMS-QTOF mass spectrometers, such as the timsTOF Pro, is complemented by robustness and reproducibility due to a combination of the low sample amounts required for analysis and the orthogonal geometry of the ion optics. This is incredibly important for the in-depth profiling of complex protein mixtures, such as human tissues or body fluids in clinical proteomics [3]. The PASEF method further increases the chance of identifying low abundance proteins, for example in blood plasma.
Conclusion
MS-based proteomics has provided an abundance of important insights into the composition, regulation and function of molecular pathways, for a deeper understanding of complex biological processes. New technological innovations, such as the PASEF method for MS-based proteomics, will continue its positive impact across biology and medicine, by its precise identification and quantification of thousands of proteins from complex samples.
Future applications of MS-based proteomics could further the field of personalized medicine, by potentially enabling clinicians to classify patients into treatment resistant and non-resistant groups, therefore advancing the application of targeted therapy. The measurement of disease-relevant biomarkers could also be developed in the future with MS-based proteomics, especially using TIMS-QTOF MS with PASEF.
For more information on Bruker’s timsTOF™ Pro powered by PASEF, please visit https://www.bruker.com/products/mass-spectrometry-and-separations/lc-ms/o-tof/timstof-pro/overview.html
References
1. Joshua A. Silveira, Mark E. Ridgeway, Frank H. Laukien, Matthias Mann, Melvin A. Park (2017) Parallel accumulation for 100% duty cycle trapped ion mobility-mass spectrometry, International Journal of Mass Spectrometry, Vol 413, pp 168-175. http://dx.doi.org/10.1016/j.ijms.2016.03.004
2. Florian Meier, Scarlet Beck, Niklas Grassl, Markus Lubeck, Melvin A. Park, Oliver Raether and Matthias Mann (2015) Parallel Accumulation−Serial Fragmentation (PASEF): Multiplying Sequencing Speed and Sensitivity by Synchronized Scans in a Trapped Ion Mobility Device, Journal of Proteome Research, Vol 14, pp 5378-5387. https://pubs.acs.org/doi/10.1021/acs.jproteome.5b00932
3. Scarlet Beck (2016) Novel quadrupole time-of-flight mass spectrometry for shotgun proteomics. PhD Thesis, Ludwig-Maximilians-Universität, München.

