Bacterial Chaperone Structure Disentangled

Complete the form below to unlock access to ALL audio articles.
When proteins are exposed to stresses like a heat shock, they lose their native structure and form toxic insoluble aggregations. Bacterial molecular chaperone ClpB and its yeast homologue Hsp104 have an ability to disentangle and reactivate the aggregated proteins.
ClpB forms a hexameric ring structure and its protomer consists of two ATPase cores, AAA1 and AAA2, and two additional domains, N-domain and M-domain. Using the chemical energy of ATP hydrolysis, the ring threads the aggregated proteins through its central pore to disentangle them. The disaggregation activity of ClpB is regulated by the rod-shaped M-domains surrounding the periphery of ClpB ring. However, the molecular-level mechanisms, such as how ATP binding and hydrolysis change the ClpB structure and how the changes induce disaggregation, are not known.
Although the three-dimensional structure of ClpB/Hsp104 has been determined by X-ray crystallography and cryo-electron microscopic single-particle analysis, information about the dynamics of individual molecules were required to understand the mechanism.

Figure 1: Shapes of ClpB rings
By using a high-speed atomic force microscopy (HS-AFM), Uchihashi and co-workers succeeded in observing structural changes of ClpB molecules with a 100-ms temporal resolution for the first time. In the presence of ATP, ClpB forms hexameric closed- and open-ring (Fig. 1).
The number of subunits in the ring and the polymorphism of the ring structures were further confirmed by native mass spectrometry and negative-staining electron microscopy analysis, respectively. During the HS-AFM observation, these two conformations converted each other, and the higher the ATP concentration, the greater the population of the closed ring. In addition, closed rings were further classified into "round" whose height was almost uniform, "spiral" in which the height changed continuously like a spiral staircase, and "twisted-half-spiral" in which two half-spiral structures faced each other (Fig. 1).
The twisted-half-spiral conformation suggested that the hexameric ring consisted of two trimers. This was also supported by sedimentation velocity analytical ultracentrifugation. These three conformations also converted each other, and it turned out that the higher the ATP concentration, the higher the frequency of the conversions. These observations revealed that the ATP binding induced the closed ring formation and its hydrolysis caused significant structural changes between the round, the spiral, and the twisted-half-spiral conformations.
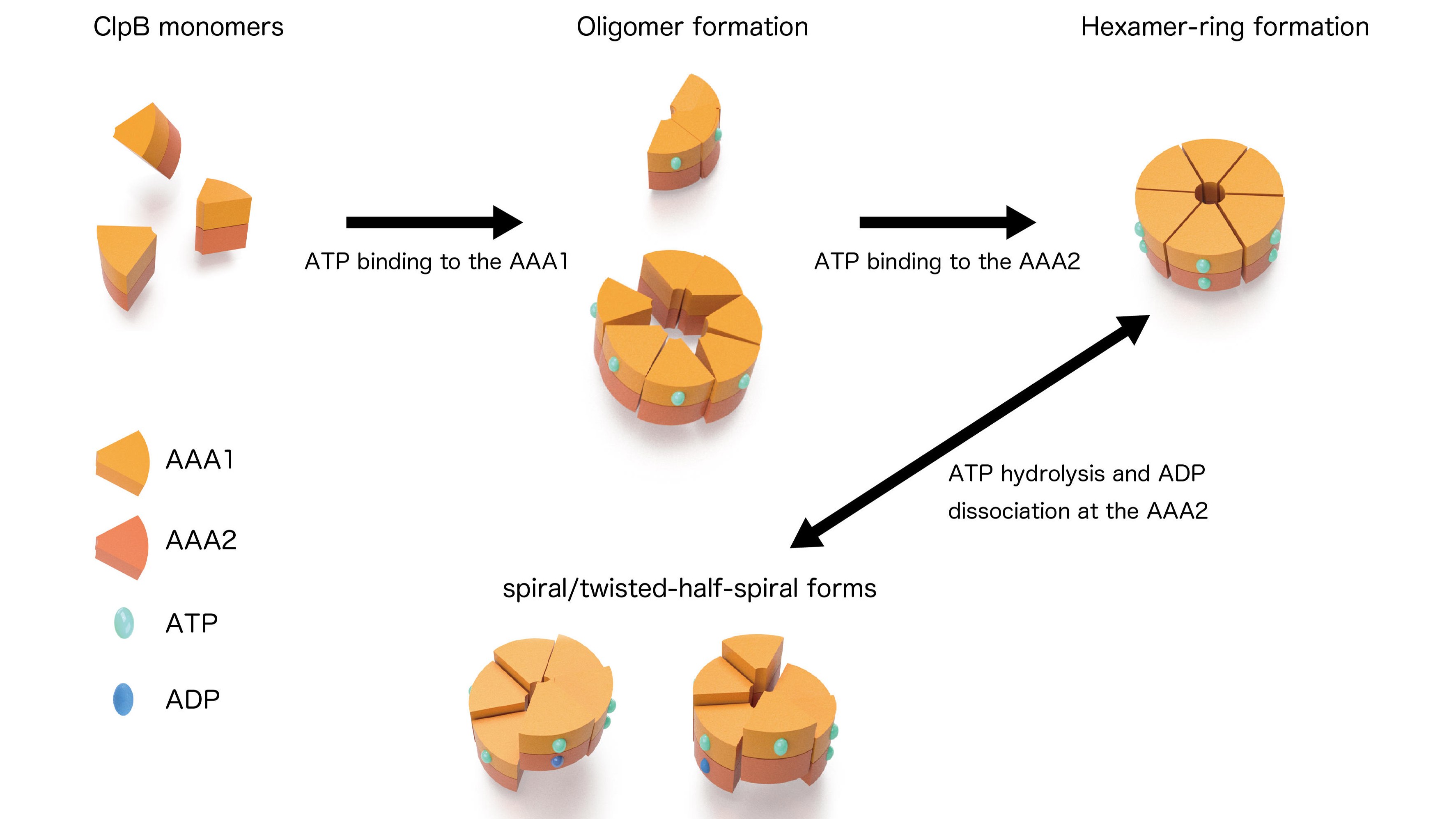 Figure 2: Dynamic changes of ClpB
Figure 2: Dynamic changes of ClpB
From the observations of ClpB mutants that were inhibited ATP binding or hydrolysis on the AAA1 and/or the AAA2, individual roles of these two domains on the structural dynamics were clarified. ATP binding to the AAA1 induces oligomerization of ClpB, and the hexameric state was stabilized by ATP binding to the AAA2. The structural changes between the round, the spiral, and the twisted-half-spiral forms were caused by ATP hydrolysis at AAA2 (Fig. 2). Moreover, the mutual structural changes of the closed rings were drastically decreased in an M-domain mutant that lost disaggregation activity but retained ATPase activity suggesting that the structural changes played an important role in the disaggregation reaction.
Protein aggregation is closely related to various diseases including Alzheimer's disease. The formation of protein aggregation is also problematic in the use of proteins in medical and industrial fields. The results of this research have a potential to contribute to treatment of such diseases and/or maintenance of useful proteins. Furthermore, ClpB belongs to the AAA+ protein family that contains various important proteins contributing to such as DNA replication, membrane fusion, protein degradation, and circadian clock maintenance. The members of this family shared AAA+ domains as ATPase cores, and the results of this research can be expected to lead to elucidation of the common mechanism of these AAA+ family proteins.
This article has been republished from materials provided by the Institute for Molecular Sciences. Note: material may have been edited for length and content. For further information, please contact the cited source.
Reference: Uchihashi, T., Watanabe, Y., Nakazaki, Y., Yamasaki, T., Watanabe, H., Maruno, T., … Ando, T. (2018). Dynamic structural states of ClpB involved in its disaggregation function. Nature Communications, 9(1), 2147. https://doi.org/10.1038/s41467-018-04587-w

