New Mouse Model Replicates Rare Neurological Disease
Complete the form below to unlock access to ALL audio articles.
A new mouse model for Joubert Syndrome has been developed by University of Bath scientists, who hope it will accelerate research to understand how the disease develops as well as help develop and evaluate therapeutic approaches.
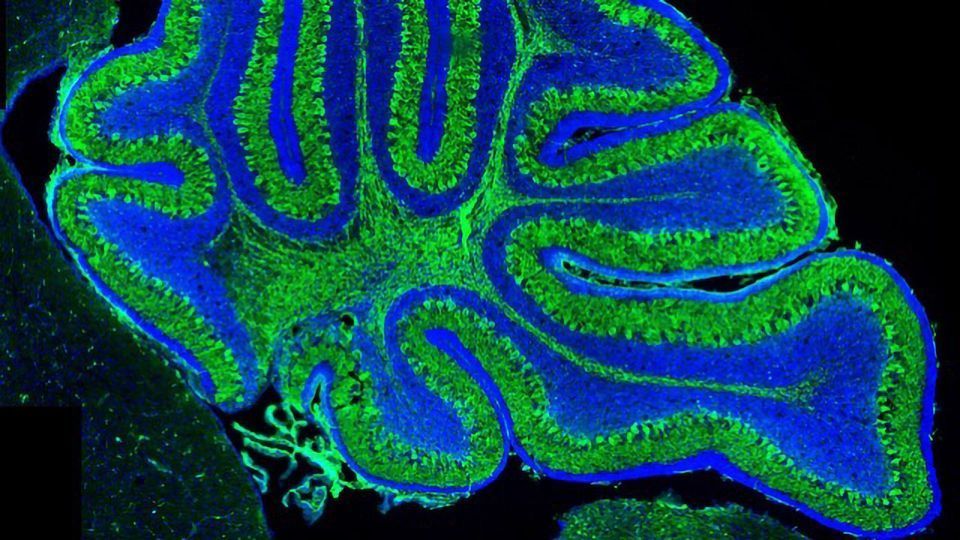
Researchers from the Department of Biology & Biochemistry were able to accurately recreate the syndrome in mice through targeted deletion of portions of a gene called Talpid3 which is required for the formation of cilia – small hair-like projections seen in many mammalian cells. These projections function like cellular antenna sensing external signals. The equivalent human gene, KIAA0856 had been linked to the disease in previous studies.
The selective gene manipulation in Talpid3 reproduced the physical abnormalities of the disease in the cerebellum of the mice. The mutant mice also demonstrated one of the key symptoms of Joubert Syndrome - progressive ataxia which is a worsening lack of motor control. In addition, the deletion in Talpid3 impacted on some key molecular pathways associated with the formation of the cerebellum.
Joubert Syndrome is a rare hereditary genetic disease, affecting between 1 in 80,000-100,000 people. The disease leads to the underdevelopment of the cerebellum, a region of the brain which controls balance and motor skills. Patients suffer from a range of symptoms, most commonly impaired motor control, abnormal breathing and sleep, and developmental delays as well as deformities such as cleft palate or extra fingers and toes.
Dr Vasanta Subramanian, who conducted the research with Dr Andrew Bashford, said: “We believe that our mouse model represents the best replication of Joubert Syndrome to date, and therefore will be an excellent experimental model for scientists studying this disease, as well in the search for new therapies.
“By selectively targeting the Talpid3 gene we have reproduced the characteristic brain abnormalities of Joubert Syndrome, as well as the symptoms, like ataxia. We believe that the deletion of Talpid3 affects signalling and cell migration in the brain, which disrupts the cellular organisation of the cerebellum, which is something we will continue to investigate in our research.”
This article has been republished from materials provided by the University of Bath. Note: material may have been edited for length and content. For further information, please contact the cited source.
Reference: Bashford, A. L., & Subramanian, V. (n.d.). Mice with a conditional deletion of Talpid3 (KIAA0586) - a model for Joubert Syndrome. The Journal of Pathology, 0(ja). https://doi.org/10.1002/path.5271

