Recovering Lost Information in Single-Cell RNA-Sequencing
Complete the form below to unlock access to ALL audio articles.
Sequencing RNA from individual cells can reveal a great deal of information about what those cells are doing in the body. MIT researchers have now greatly boosted the amount of information gleaned from each of those cells, by modifying the commonly used Seq-Well technique.
With their new approach, the MIT team could extract 10 times as much information from each cell in a sample. This increase should enable scientists to learn much more about the genes that are expressed in each cell, and help them to discover subtle but critical differences between healthy and dysfunctional cells.
"It's become clear that these technologies have transformative potential for understanding complex biological systems. If we look across a range of different datasets, we can really understand the landscape of health and disease, and that can give us information as to what therapeutic strategies we might employ," says Alex K. Shalek, an associate professor of chemistry, a core member of the Institute for Medical Engineering and Science (IMES), and an extramural member of the Koch Institute for Integrative Cancer Research at MIT. He is also a member of the Ragon Institute of MGH, MIT and Harvard and an institute member of the Broad Institute.
In a study appearing this week in Immunity, the research team demonstrated the power of this technique by analyzing approximately 40,000 cells from patients with five different skin diseases. Their analysis of immune cells and other cell types revealed many differences between the five diseases, as well as some common features.
"This is by no means an exhaustive compendium, but it's a first step toward understanding the spectrum of inflammatory phenotypes, not just within immune cells, but also within other skin cell types," says Travis Hughes, an MD/PhD student in the Harvard-MIT Program in Health Sciences and Technology and one of the lead authors of the paper.
Shalek and J. Christopher Love, the Raymond A. and Helen E. St. Laurent Professor of Chemical Engineering and a member of the Koch Institute and Ragon Institute, are the senior authors of the study. MIT graduate student Marc Wadsworth and former postdoc Todd Gierahn are co-lead authors of the paper with Hughes.
Recapturing information
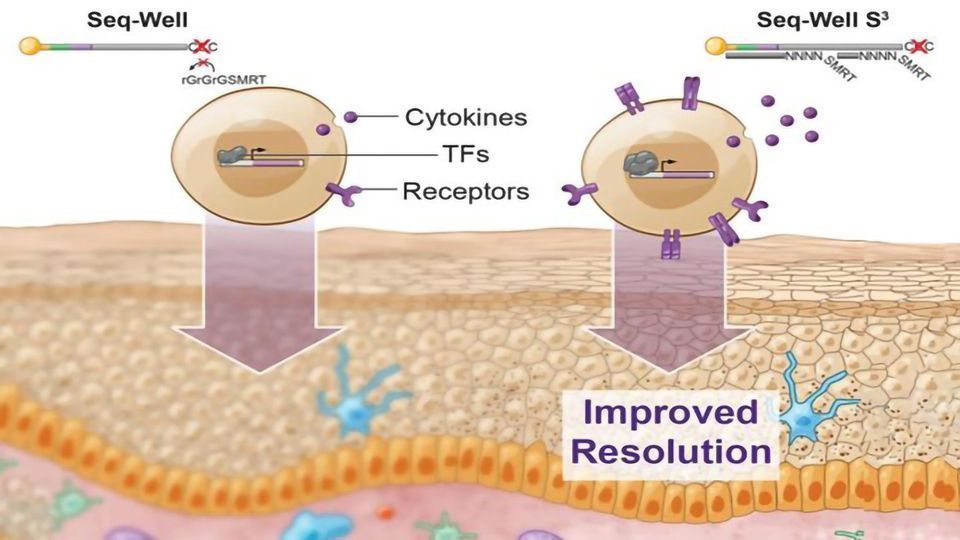
A few years ago, Shalek, Love, and their colleagues developed a method called Seq-Well, which can rapidly sequence RNA from many single cells at once. This technique, like other high-throughput approaches, doesn't pick up as much information per cell as some slower, more expensive methods for sequencing RNA. In their current study, the researchers set out to recapture some of the information that the original version was missing.
"If you really want to resolve features that distinguish diseases, you need a higher level of resolution than what's been possible," Love says. "If you think of cells as packets of information, being able to measure that information more faithfully gives much better insights into what cell populations you might want to target for drug treatments, or, from a diagnostic standpoint, which ones you should monitor."
To try to recover that additional information, the researchers focused on one step where they knew that data was being lost. In that step, cDNA molecules, which are copies of the RNA transcripts from each cell, are amplified through a process called polymerase chain reaction (PCR). This amplification is necessary to get enough copies of the DNA for sequencing. Not all cDNA was getting amplified, however. To boost the number of molecules that made it past this step, the researchers changed how they tagged the cDNA with a second "primer" sequence, making it easier for PCR enzymes to amplify these molecules.
Using this technique, the researchers showed they could generate much more information per cell. They saw a fivefold increase in the number of genes that could be detected, and a tenfold increase in the number of RNA transcripts recovered per cell. This extra information about important genes, such as those encoding cytokines, receptors found on cell surfaces, and transcription factors, allows the researchers to identify subtle differences between cells.
"We were able to vastly improve the amount of per cell information content with a really simple molecular biology trick, which was easy to incorporate into the existing workflow," Hughes says.
Signatures of disease
Using this technique, the researchers analyzed 19 patient skin biopsies, representing five different skin diseases -- psoriasis, acne, leprosy, alopecia areata (an autoimmune disease that causes hair loss), and granuloma annulare (a chronic degenerative skin disorder). They uncovered some similarities between disorders -- for example, similar populations of inflammatory T cells appeared active in both leprosy and granuloma annulare.
They also uncovered some features that were unique to a particular disease. In cells from several psoriasis patients, they found that cells called keratinocytes express genes that allow them to proliferate and drive the inflammation seen in that disease.
The data generated in this study should also offer a valuable resource to other researchers who want to delve deeper into the biological differences between the cell types studied.
"You never know what you're going to want to use these datasets for, but there's a tremendous opportunity in having measured everything," Shalek says. "In the future, when we need to repurpose them and think about particular surface receptors, ligands, proteases, or other genes, we will have all that information at our fingertips."
The technique could also be applied to many other diseases and cell types, the researchers say. They have begun using it to study cancer and infectious diseases such as tuberculosis, malaria, HIV, and Ebola, and they are also using it to analyze immune cells involved in food allergies. They have also made the new technique available to other researchers who want to use it or adapt the underlying approach for their own single-cell studies.
Reference: Hughes TK, Wadsworth MH II, Gierahn TM, et al. Second-strand synthesis-based massively parallel scRNA-Seq reveals cellular states and molecular features of human inflammatory skin pathologies. Immunity. 2020;53(4):878-894.e7. doi:10.1016/j.immuni.2020.09.015.
This article has been republished from the following materials. Note: material may have been edited for length and content. For further information, please contact the cited source.

