

MetaboScape - Providing a new layer of Insight in Discovery Metabolomics
Discovery metabolomics research workflows require the ability to quickly pinpoint and confidently identify relevant markers in cohorts of complex mixtures. By enabling these workflows and providing options to use pathway mapping to set results in a biological context MetaboScape turns complementary data from Brukers LC-QTOF-MS/MS, and Magnetic Resonance Mass Spectrometry (MRMS) into knowledge.
Data pre-processing is an important step to provide confidence in statistical results in non-targeted Metabolomics. Missing values can cause false negative results in statistical data evaluation - real differences present in the data are not highlighted as being different. The Time aligned Region complete eXtraction (T-ReX) algorithm extracts all relevant information, automatically and in a “region complete” manner. In cohorts of LC-QTOF-MS/MS data sets ions belonging to the same compound are combined, aligned across all samples and automatically re-extracted in individual samples if initially below the peak picking threshold therefore addressing the missing value problem in statistics.
1497004903587.png)
1497453195626.png)
Figure 1: The T-ReX algorithm automatically recalibrates LC-MS/MS data from Bruker QTOF instruments (A 1). Subsequently T-ReX combines ions belonging to the same compound into one feature, i.e. isotopes, charge states, adducts or fragments (A 2). Non-linear retention time alignment ensures data consistency in large cohorts of LC-QTOF-MS data sets (B 1). Features are extracted “region complete” across multiple samples addressing the “missing values” problem for subsequent statistical analysis (B 2).
Automatic and confident identification of known compounds is essential to fully understand the biological context of metabolomics data. The graphical Annotation Quality “AQ” representation enables the analyst to readily evaluate the reliability of each annotation automatically generated in MetaboScape by matching retention time, accurate mass, isotopic pattern information, and MS/MS spectral library spectra according to user definable threshold levels.

Figure 2: Graphical Annotation Quality “AQ” symbol enables the analyst to readily evaluate their confidence in automatically identified compounds
MetaboScape also supports flow-injection (FI) or MALDI- Magnetic Resonance Mass Spectrometry (MRMS), two revolutionary extreme mass resolution techniques that enable higher sample throughput for profiling complex metabolic extracts. The intuitive workflow for evaluating this LC free data provides access to compounds not readily addressed by complementary LC-MS analysis.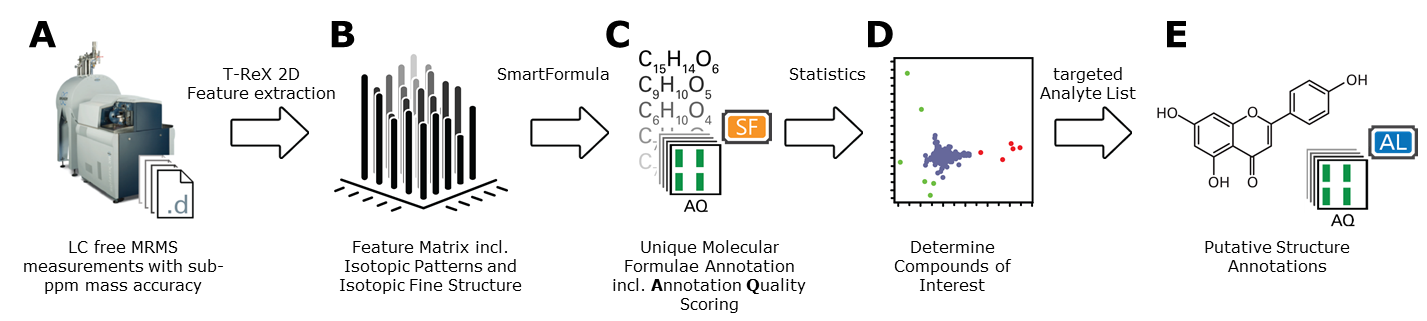
Figure 3: Schematic representation of the FI / MALDI-MRMS workflow supported in MetaboScape 3.0: A) LC free MRMS acquisition; B) Creation of a feature matrix, where a feature comprises the pseudo molecular ions and isotopologues of putative molecules; C) A list of molecular formula annotations, including Annotation Qualities scoring; D) Statistical analysis (e.g. PCA) to identify features of interest; E) Putative structure annotations based on unique molecular formula.





