The Evolution of Proteomics - Dr Richard Scheltema

Complete the form below to unlock access to ALL audio articles.
Richard Scheltema is an assistant professor at Utrecht University where he heads up the Scheltema laboratory within the wider context of the group of Albert Heck.
The research group focuses on mass spectrometry (MS) based structural proteomics, for which they develop advanced liquid chromatography-mass spectrometry (LC-MS/MS) platforms and analysis software, working closely with other researchers such as Alexander Makarov who features later in The Evolution of Proteomics.
Scheltema is the core developer of XlinkX for Proteome Discoverer, and with his team adopts this technology to gain an in-depth quantitative view on proteins, in addition to gathering special information to answer interesting biological questions.
Molly Campbell (MC): What has been the greatest breakthrough in the proteomics research field in recent years?
Richard Scheltema (RS): To me, the applications that MS based proteomics have seen over the years are very exciting. An instrument that is basically only measuring masses and rough abundances is capable of:
- extracting the presence of proteins
- extracting which post-translational modifications are present on which proteins and in which stoichiometry
- estimating copy-numbers
- uncovering which proteins are interacting
- deriving structural information
- figuring out the cellular localization of proteins
MC: Your research utilizes cross-linking mass spectrometry (XL-MS). Can you tell us more about this approach and why, for your work, it is superior to other available techniques?
RS: In XL-MS we are interested in investigating protein structure and protein-protein interactions. This is achieved at the "crosslinking" stage by solubilizing the protein (-complex) in its native state and incubating it with crosslinking reagents – small chemicals with two amine reactive ends that form a covalent bond between two amino acids. After the crosslinking stage, the protein(-complex) is processed which finally sees the protein(s) cut into peptides by a protease (Figure 1, panel I).

This results in a sample containing normal peptides and copies of 2 peptides connected by the crosslinking reagent. This mixture is subsequently measured by MS for identification – where in most cases the amino acids involved in the crosslink can be assigned providing us with a distance constraint defined by the length of the spacer arm and the two side chains. These distance constraints provide valuable information on how the protein is folded (two peptides originating from the same protein) or on which proteins are interacting and where the interface of this interaction is located (two peptides originating from different proteins).
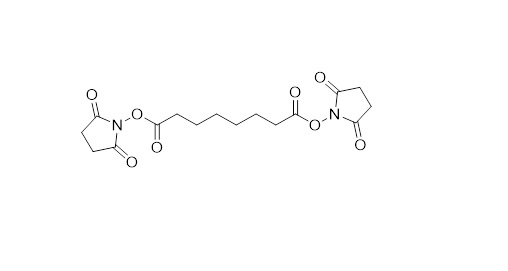
The reactive ends are separated by a spacer arm and typically the sidechains of the amino acids are targeted, resulting in a structural resolution between 15 and 50 Å (Figure 2). This is by no means near the resolution that one can achieve with techniques such as Crystallography, electron microscopy (EM), or nuclear magnetic resistance (NMR) spectroscopy. Therefore, we do not aim to compete with these techniques for resolving structural information – I rather see what we do as highly complementary. We are for example not limited in size of the proteins and protein-complexes under investigation, can peak inside the protein structure, are not bothered by flexibility in the protein structure and can deal with highly complex mixtures. Even though the information is of relatively low resolution, when combined with high resolution crystal structures of individual subunits and/or high-resolution EM maps, a very detailed structural picture can be built up.

Figure 3: XL-MS combined with Cryo-TEM. Original figure. Credit: Richard Scheltema.
Our ability to tackle complex lysates will most likely result in XL-MS gaining a lot of traction in the foreseeable future.
Transmission electron cryomicroscopy (Cryo-TEM), where whole cells are imaged by EM, is seeing a great surge in use. From the protein structural outlines recorded with this technique, it is difficult to identify which proteins are involved with each other and how they are arranged within the full structure – questions we are providing answers to.
With this in mind, we applied for a grant at the Dutch Science Council with the goal of developing approaches to marry Cryo-TEM with XL-MS. We hope to achieve this through the use of automated protein docking solutions where we want to infuse a lot of knowledge gained from the MS measurements. We are very confident that we will be able to succeed and we aim to investigate organisms with a large potential for discovering new protein complexes. The project is led by an excellent team of principal investigators, such as John van der Oost, Albert Heck, Alexandre Bonvin, Friedrich Föerster, and myself.
RS: Proteomics data processing software is one of the focusses of my laboratory in the larger context of the Heck group. The samples recorded in XL-MS studies have an extra level of complexity as we record data with two still connected peptides (in contrast to normal proteomics experiments where a single peptide is measured). When we started developing our software, solutions were already available; however, we wanted to step into this field to enable experiments with highly complex lysates – an area at that point then not yet covered by existing solutions. In addition, we found the ability to change the data analysis software also highly beneficial for the flexibility it offers to perform "out-of-the-box" experiments. During development, besides ensuring correctness of the extracted peptide identifications, we placed emphasis on user friendliness. We defined this as (1) ability to run on any desktop pc (which requires a large amount of optimization of the algorithms), (2) easy presentation of the results, requiring graph visualizations and browsable tables and (3) support in case of questions and/or problems.

MC: In 2018 you published a study in which your team looked at histone protein interaction landscapes using XL-MS in intact cell nuclei. Can you tell us about your findings and what they contribute to the field? How can further research expand the data?
RS: Crosslinking of the intact nucleus provided a snapshot of the histone interaction network and set the basis for investigations into how stimuli influence chromatin organization and influence/regulate the histone interactome. However, further experiments – and importantly, increased depth of analysis over what we achieved – are needed. We would like to apply our nuclear XL-MS workflow in cells treated with histone deacetylase inhibitors (HDAC inhibitors) that promote unpacking of chromatin. This relaxed state of chromatin promotes transcription and will allow us to investigate the dynamics and organization of endogenous transcriptional complexes.
What we have already in part uncovered is how repressive and activating histone marks (methylation and acetylation of histone tails) drives interactions to other proteins. Additionally, we uncovered for known interactors the interaction interfaces to the histones, which we could model on the existing structure of histone.
MC: What are some of the key challenges you face in structural proteomics?
RS: There are two major concerns for XL-MS experiments. The first has to do with the low abundance of the crosslinked peptide products. From available data, we estimate that over 99% of the material injected into the mass spectrometer consists of normal peptides (i.e. peptides not modified by the crosslinking reagent), carrying no structural information.
Another approach researchers have been taking is to integrate an enrichment handle directly on the crosslinking reagent – creating a so-called tri-functional crosslinking reagent. Previously, Biotin has been used as the handle. The produced reagents however never found traction as it remains difficult to efficiently detach Biotin from the beads used for capture. The Biotin handle additionally makes the reagent very bulky potentially leading to steric hindrance and low access into the protein structure under investigation.
Our solution to this problem – and we really think this is now resolved – was to take a cue from the developments in phosphorylated peptide enrichment with immobilized metal affinity chromatography (IMAC) technologies. The advantage of IMAC is that it has seen large scale automation, phospho-groups can be easily detached from the beads, it has fantastic specificity (meaning non-phosphorylated peptides can be almost completely separated from the phosphorylated ones) and importantly the phospho-group is very small in comparison to Biotin. To this end, we developed a tri-functional crosslinking reagent, PhoX, that incorporates a phospho enrichment handle. From our experiments we found that with this reagent we indeed get incredible performance in creating pure samples and thereby making the detection of crosslinked peptides very easy.
The second major concern for these experiments is that we are able to generate a lot of structural data which currently has no protein data bank structure associated to it. Especially from complex lysates we observe many complexes of which the individual subunits never were (successfully) crystalized or recorded with EM. This essentially prevents us from building up a structural picture of the complexes based on crosslinks between the individual subunits of the complex. To deal with this situation we and others revert to structural modelling where we use the detected intra-links for each of the individual subunits. The developed tools available are really fantastic as they are able to provide a structure for a given amino acid sequence that is, in a lot of cases, very reasonable. The problem is that we tend to have to search for the best structure in a pool of many generated possibilities. For this, the detected crosslinks are very helpful in validating and filtering, but a large amount of manual work is also required to extract the biologically most relevant model from all the possibilities. This leads to very protracted time-frames for projects trying to uncover structural details. For this, so far no easy fixes have been proposed and the hunt remains ongoing.
RS: The obvious advancement would of course be further improvements in the MS platforms we utilize, but also the liquid chromatography platforms we use to separate the peptide mixtures over a single measurement would be very much appreciated – both in the terms of performance and stability, but particularly stability remains a point of some concern. Also, the development of novel fragmentation techniques and improvements to existing approaches would be very nice to see.
Finally, I would be very excited to see advances in protein purification, sample preparation, data analysis software - basically every aspect of proteomics. All information generated is useful for our structural studies, so, everything is welcome!


