The Give and Take Relationship of XL-MS and Cryo-EM

Complete the form below to unlock access to ALL audio articles.
Structural biology has recently observed the marrying of two remarkably complimentary techniques for the study of protein structure and interaction: cross-linking mass spectrometry (XL-MS) and the Nobel prize winning cryogenic electron microscopy (cryo-EM). As a "power" couple, they work together to overcome their individual flaws. When molecular regions are less well defined in cryo-EM images, XL-MS steps in to provide critical information on the proximity of specific amino acid residues, allowing identification of the protein and an accurate deduction of protein structure. Here, we take a closer look at the evolution of these techniques and how, together, they look set to propel the field of structural proteomics into a new era.
The blueprint of an assembled machine
Proteins and their complexes are the biological "workhorse" of cells, regulating processes integral to cell function such as cell growth, cell death and each stage of the cell's lifecycle in between.
"I like to compare a protein structure to a blueprint of an assembled machine," says Cristina Paulino, Assistant Professor in high-resolution cryo-EM at the University of Groningen, in a recent interview. "Whilst genetics and biochemistry help in understanding what the physiological role of a protein is, structural biology uncovers what these nanomachines look like and how they are wired."
Knowledge of this "wiring" therefore presents scientists the opportunity to repair proteins, engineer and replicate them or potentially block their function – applications of proteomics that are forecast to be integral to personalized medicine and modern pharmacology.
XL-MS a key component of the MS toolbox
A basic principal in biology is that proteins consist of amino acid residues linked by peptide bonds to form polypeptides. In addition to the peptide bonds, non-covalent bonds, such as van der Waals forces, electrostatic and hydrophobic interactions also exist. In structural biology, these bonds are hard to detect, adding an additional layer of complexity when studying protein structure at the atomic level. In the past decade, the proteomics field has witnessed an impressive array of technologies added to the MS toolbox. XL-MS is one such technology that has proven indispensable to structural proteomics.1
Figure 1 summarizes a typical XL-MS workflow, in which non-covalent interactions between proteins (or in their proximity) are converted to artificial covalent bonds by solubilizing the native protein with a crosslinking reagent. Due to its prevalence, stability in aqueous solutions and high reactivity, the primary amine group of lysine residues or a protein's N-terminus is a frequent target for cross-linkers. Here, homobifunctional cross-linkers such as disuccinmidyl suberate (DSS) and bis(sulfosuccinimidyl) suberate are most commonly adopted.2 After the crosslinking stage, the protein is processed and cut into peptides by a protease. 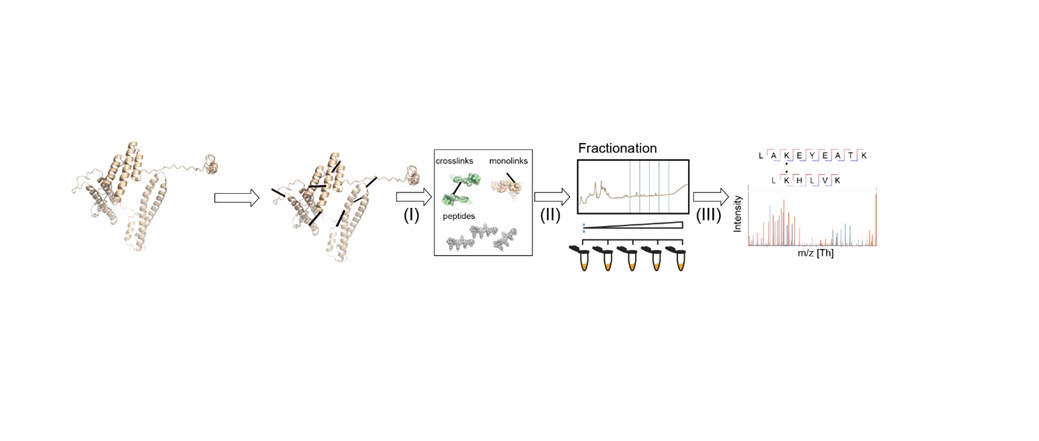 Figure 1: General XL-MS workflow. Credit: Richard Scheltema, Heck Laboratory, Utrecht University.
Figure 1: General XL-MS workflow. Credit: Richard Scheltema, Heck Laboratory, Utrecht University.
"The mixture is subsequently measured by MS for identification – where in most cases the amino acids involved in the crosslink can be assigned providing us with a distance constraint defined by the length of the spacer arm and the two side chains," explains Richard Scheltema, PhD, from the Heck Laboratory at Utrecht University. "These distance constraints provide valuable information on how the protein is folded (two peptides originating from the same protein) or on which proteins are interacting and where the interface of this interaction is located (two peptides originating from different proteins)."
Typically, the structural resolution achieved by XL-MS is between 15 and 50 Å – no match for the resolutions attained by alternative structural biology techniques such as X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy and of course, cryo-EM, hence why the techniques must be used in complement with each other.3
Cryo-EM offers a reSOLUTION
Emerging from transmission electron microscopy (TEM), cryo-EM determines a three-dimensional (3D) structure from a two-dimensional (2D) image projection, whilst maintaining the integrity and the structure of the sample in a close-to-native state. This is achieved by studying the sample in a vitrified state, whereby a thin slide of the sample is rapidly plunged into a liquid ethane bath, cryogenically preserving and protecting it from the vacuum within the TEM and radiation damage.4 Paulino discusses the advantages of adopting cryo-EM over other structural biology techniques further here.
The cross-link data in XL-MS describes the maximum distance between two specific amino acid residues in a peptide. Suggested structural models of proteins and their domains are slotted into the reconstituted 3D volume obtained through cryo-EM, and the cross-linking data is integrated to validate the location and orientation of specific peptides within the protein complex.
Recently, Henry et al. determined the active structure and binding mechanism of apolipoprotein E4 (ApoE4). Implicated in Alzheimer's disease (AD) and cardiovascular disease (CVD), ApoE4 is a lipidated isoform of apolipoprotein (ApoE), a protein that facilitates the internalization of cholesterol rich lipoproteins by acting as a ligand for cell surface receptors. Adopting a hybrid approach incorporating XL-MS, cryo-EM and bioinformatic modeling tools, Henry et al. showed that ApoE4 exists in two alternate confirmations, pointing towards an activation mechanism that relies on regulation of the accessibility of its receptor binding region. The authors note that such findings could prove valuable to interpreting the role of the protein in AD and CVD and subsequently the development of potential therapeutics.6
Similar impressive efforts are outlined by Schmidt and Urlaub in their comprehensive 2017 review, including the structural characterization of the spliceosome by the Lührmann and Stark groups.3
In January of this year, the Netherlands Organization for Scientific Research (NWO) awarded a €1.6 million euro grant to a project titled Monitoring and Visualizing Protein Societal Behavior in the Cell in which XL-MS and cryo-EM techniques, amongst other molecular methods, were used collectively. The project sees an established team of principal investigators, including Albert Heck, John van der Oost, Alexandre Bonvin, Friedrich Föerster, and Scheltema map and visualize interactions between proteins.
"In this project we aim to use cryo-tomography, a specialized application of cryo-EM, to unbiasedly uncover all protein complexes in a select set of thermophiles. XL-MS is used here to provide identities and spatial ordering of the proteins inside the identified complexes (something which typically cannot be directly answered from the tomography data) and structural modelling to fill in the final gaps." Says Scheltema "The thermophiles were selected as these organisms are a potential treasure trove for protein complexes with biochemical use."
Redefining limitations and progressing forward
Together, XL-MS and cryo-EM offer the potential to significantly advance the structural proteomics field. However, they each face their own individual limitations that must be overcome to form a perfect partnership.
"Cryo-EM is continuously redefining its limitations, however we still face several challenges," comments Paulino. "Whereas for X-ray crystallography, access to fully operational and maintained synchrotron beam lines is largely available and free, the overall costs in cryo-EM and the level of expertise required to operate the microscopes is an obstacle. This has been, to some extent but not entirely, addressed by the implementation of government-subsidised cryo-EM facilities".
"Cryo-tomography provides a comparatively low-resolution view on the protein complexes from which direct interpretation is difficult." Scheltema adds "The data from XL-MS on the other hand provides a view of everything that exists in solution with spatial information. However, linking these two provides I think the biggest challenge because XL-MS delivers information on all proteins in the sample, which needs to somehow be filtered for proteins inside the complexes unveiled by tomography."
References:
1. Rappsilber, Juri. 2011. The Beginning of a Beautiful Friendship: Cross-Linking/Mass Spectrometry and Modelling of Proteins and Multi-Protein Complexes. Journal of Structural Biology. https://doi.org/10.1016/j.jsb.2010.10.014.
2. Yu and Huang. 2017. Cross-Linking Mass Spectrometry (XL-MS): An Emerging Technology for Interactomics and Structural Biology. Analytical Chemistry. https://doi.org/10.1021/acs.analchem.7b04431.
3. Schmidt and Urlaub. 2017. Combining Cryo-Electron Microscopy (Cryo-EM) and Cross-Linking Mass Spectrometry (CX-MS) for Structural Elucidation of Large Protein Assemblies. Current Opinion in Structural Biology. https://doi.org/10.1016/j.sbi.2017.10.005.


