

Why Does the Molecular Weight of My Protein Differ From the Theoretically Expected Weight?

Complete the form below to unlock access to ALL audio articles.
Western blotting and molecular weight calculations
Western blotting (or immunoblotting) is a commonly used molecular biology technique for analyzing proteins. The predicted protein molecular weight (MW) is the sum of all protein amino acid MWs. It can be calculated using, for example, the online ExPASy tool. However, the calculated MW may be different from that observed on the Western blot. The figure below summarizes the most common reasons as to why this may occur (Figure 1).

Figure 1. The most common reasons for differences between observed and theoretical MWs. Credit: Proteintech.
1. The signal peptide (and a pro-peptide) gets cleaved off
Many proteins that are transported through the secretory pathway have signal peptides of 15–35 amino acids in length, located predominantly at their N-termini. They are often cleaved by various proteases during subcellular transport, resulting in the mature protein running at a lower than predicted MW. The presence of a signal peptide can be predicted using various online tools or may be established based on previously published data. They are usually well annotated in protein databases, e.g., UniProt. Additionally, a subset of proteins has pro-peptides – protein domains that are present in protein precursors. Protein precursors need to be processed by proteases in order to engender a functional product, without a pro-peptide (Figure 2). Figure 2. PINK1 (23274-1-AP) is a mitochondrial serine/threonine-protein kinase that protects cells from stress-induced mitochondrial dysfunction. The precursor of PINK1 (65 kDa) is synthesized in the cytosol and is imported into the outer membrane of mitochondria. PINK1 is further transferred into the inner membrane where it is cleaved into a 52 kDa mature form.
Figure 2. PINK1 (23274-1-AP) is a mitochondrial serine/threonine-protein kinase that protects cells from stress-induced mitochondrial dysfunction. The precursor of PINK1 (65 kDa) is synthesized in the cytosol and is imported into the outer membrane of mitochondria. PINK1 is further transferred into the inner membrane where it is cleaved into a 52 kDa mature form.
Caspases, a family of endoproteases, are critical players in cell regulatory networks controlling inflammation and cell death. Caspase 3 (19677-1-AP) exists as an inactive proenzyme form of 32 kDa (p32), which, upon apoptotic signaling, is cleaved into two active subunits (p19/17 and p12) that assemble into a functional tetrameric enzyme (PMID: 7596430).
Proteins of the matrix metalloproteinase (MMP) family are involved in the breakdown of the extracellular matrix in many physiological processes, including embryonic development, reproduction, tissue remodeling, and disease processes such as arthritis or metastasis. Most MMPs are secreted as inactive pro-proteins that are activated when cleaved by extracellular proteinases. The inactive pro-MMP9 (10375-2-AP) is 92 kDa. It is sequentially cleaved by MMP3 into a processed form of 68 kDa through an intermediate form – 78/82 kDa (PMID: 1371271). MMP9 can also exist as a dimer of 180 kDa (PMID: 7492685). Credit: Proteintech.
2. Post-translational modifications (PTMs)
PTMs are covalent modifications of proteins that occur after their synthesis. Many modifications are catalyzed by enzymes that reside within the secretory pathway, i.e. the endoplasmic reticulum and the Golgi apparatus. PTMs are important regulators of protein-protein interactions, protein stability, function, enzymatic activity, and localization. The most common modification is phosphorylation, which occurs predominantly on serine, tyrosine and threonine residues. Phosphorylation, like many other PTMs, is often transient – kinases catalyze the attachment of the phosphoryl groups, while phosphatases remove them. Most proteins undergo N-terminal acetylation, an enzymatic reaction catalyzed by N-terminal acetyltransferases (NATs). Some amino acid residues can be linked with oligosaccharides in a process known as N- and O-linked glycosylation. Ubiquitination, the addition of ubiquitin, is a common initial step for proteasome degradation. You can find out more about PTMs here.
Glycosylation and glycanation
Most proteins that are synthesized on ribosomes associated with the endoplasmic reticulum undergo glycosylation. This means that a covalent attachment of sugar moieties is added to the polypeptide chain.
Phosphorylation
One of the most common PTMs is protein phosphorylation, which takes place on serine, threonine, and tyrosine residues. Phosphorylation regulates protein function, its enzymatic activity, protein–protein interactions, and protein localization.
The addition of a single phosphoryl group adds +/- 1 kDa to the MW, which is often beyond the resolution of the standard SDS-PAGE. However, multiple phosphorylation sites can lead to more prominent MW changes.
Ubiquitination
Protein ubiquitination means a covalent ubiquitin is added to lysine, cysteine, serine, threonine, or directly to the protein N-terminus. Ubiquitination via the proteome can mark proteins for degradation.
3. Protein complexes
The majority of protein complexes consist of proteins associated by non-covalent bonds. These interactions are destroyed during sample preparation and electrophoresis, and individual proteins run as monomers. This is due to the fact that SDS-PAGE for Western blotting is performed in reducing conditions. Despite this, some protein complexes are not fully disrupted, and proteins can still exist in the form of homo- or heteromeric complexes, even in the presence of reducing agents, such asSDS and β-Mercaptoethanol. The observed MW of the complexes is subsequently higher than the calculated MW of monomers. It is also common to observe multiple bands that represent monomeric and complex species (Figure 3).
Note: 20% β-Mercaptoethanol (or 100 mM DTT) for the 4X SDS sample buffer might help to remove unspecific bands due to dissociation of the protein complex.
Figure 3: NQO1 (11451-1-AP) is an enzyme that serves as a quinone reductase together with conjugation reactions of the hydroquinones involved in detoxification pathways as well as in biosynthetic processes such as the vitamin K-dependent gamma-carboxylation of glutamate residues in prothrombin synthesis. NQO1 has three isoforms: 26, 27, and 31 kDa MW, and the formation of homodimers (66-70 kDa) is needed for its enzymatic activity.
Mlx-interacting protein (MLXIP, also known as MONDOA) (13614-1-AP) acts as a transcription factor forming a heterodimer with MLX protein. This complex binds and activates transcription from CACGTG E boxes, playing a role in the transcriptional activation of the glycolytic target and glucose-responsive gene regulation. MLXIP has three isoforms: 110, 57, and 69 kDa, and the MW of the MLXIP-MLX heterodimer is 130 kDa. Credit: Proteintech.
4. Protein isoforms
In eukaryotes, newly transcribed pre-mRNAs undergo differential mRNA maturation steps, leading to mRNA species that differ in the number and length of exons – protein-coding sequences. Protein variants of the same gene are regarded as protein isoforms. Protein isoforms can differ in tissue expression patterns and they can have distinctive physiological roles. Because of their varied exon content, protein isoforms can differ in MW (Figure 4). Additionally, mRNAs can bear more than one translation start site (TSS) marking the start of the N-terminus of proteins. The use of multiple TSSs creates protein isoforms with a distinct N-terminus and hence different MW. 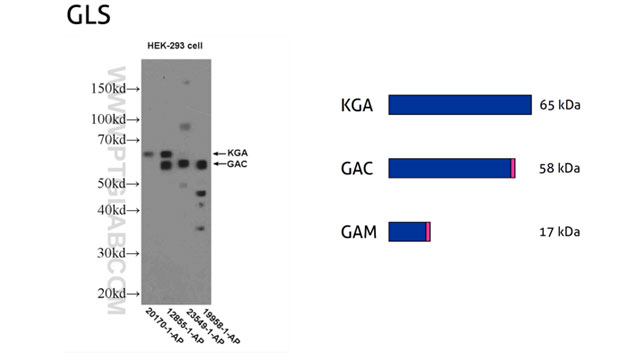
5. Technical obstacles
a. Antibody cross-reactivity
It is relatively common to observe additional reactive species during Western blotting that do not correspond to any forms of the target protein. Instead, they represent unrelated proteins that are non-specifically recognized by the antibody. Antibody cross-reactivity usually requires careful verification with appropriate controls and can be minimized through the optimization of experimental conditions.
The choice of extraction buffer is essential in decreasing the effect of antibody cross-reactivity. If the analyzed protein is cytoplasmatic, a mild extraction buffer that does not extract nuclear proteins (e.g., using saponin as a detergent) may be more suitable to lower non-specific cross-reactivity with nuclear proteins. Following the transfer of the protein from polyacrylamide gel onto a membrane, the type of blocking buffer, incubation times of membranes with primary and secondary antibodies, and washing step all require optimization. It is recommended to compare different antibody types raised against the target protein. If cross-reactivity is detected with polyclonal antibodies, it may not be seen with monoclonal antibodies that are raised against a specific epitope rather than the whole protein sequence or a larger protein fragment.
b. Unspecific proteolytic cleavage and protein degradation
Proteins can be subject to non-specific proteolytic cleavage and degradation. The breaking down of cells and tissues releases extra- and intracellular proteases that can cleave proteins into polypeptides. Therefore, it is vital to supplement lysis buffers with protease inhibitors that block the activity of proteases. A suggestion is to perform cell lysis on ice to further prevent protein degradation. Both proteolytic cleavage and degradation can affect proteins to various degrees and result in protein fragments with a lower MW.
Observed MW | Potential Causes |
| Higher than expected | 1. PTMs (see point 2). 2. Antibody is detecting a protein isoform with a longer sequence (see point 4). 3. Protein complexes (see point 3). |
| Lower than expected | 1. Cleavage of signal peptide (see point 1). 2. Antibody is detecting a protein isoform with a shorter sequence (see point 4). 3. Unspecific protein cleavage (see point 5b). |
| More than one band observed | 1. Protein isoforms (see point 4). 2. One protein product but with different posttranslational modifications (see point 2). 3. Antibody is detecting protein with and without pro-peptide (see point 1). 4. Protein complexes (see point 3). 5. Antibody cross-reactivity (see point 5a), potentially due to homology of the immunogen sequence. |
| No bands observed | 1. Protein cleavage (see point 5b). 2. Protein degradation (see point 5b). - Make sure to include appropriate controls (see point 5a). - It is possible that further protocol optimization is needed (see point 5a). |
Table 1: What is the observed MW, and what is the reason behind it?

