


Analyse ALL of your data: Don’t settle for missing values –Progenesis QI’s unique co-detection solution
Missing values are a major problem in LC-MS based discovery ‘omics analysis and can have profound implications upon the success of the project.

In discovery ‘omics, we look for differences in the relative quantities of analytes between two or more groups or conditions. In all biological systems, however, there is inherent biological variation caused by both genetic and environmental factors, hence the relative quantity of any given analyte ion will vary across samples from different specimens within a given condition.
This inherent variation can be seen as biological “noise” which we must cut through to find the consistent, condition-related differences we’re looking for. To do this, we must run multiple biological replicate samples from different specimens of the same condition, comparing the resulting sample groups to find the analyte ions that display statistically significant differences between conditions. The more biological replicate samples we run, the greater the ability or statistical power of our experiment to find the significantly changing ions.
The relationship between the number of replicate samples and the statistical power of the experiment is strongest when data from all the runs is available for statistical analysis. However, due to limitations in the conventional workflow used by most analysis software, this is usually not the case.
Indeed, using conventional but inefficient workflows, the feature ion signals are detected independently on each sample. This creates different detection patterns, even for technical replicates, so that matching the ions to ensure you are comparing ‘like with like’ across all samples becomes very difficult. This leads to the generation of many “missing values” in the ion quantity matrix. Multivariate statistical analysis is then performed on the ion quantity matrix, to find the truly significant expression changes. However, the impact of having missing values in the ion quantity matrix means that it is not actually possible to do a ‘like with like’ comparison on many features.
This means the multivariate statistics have to be applied to a restricted number of features or an imputation method has to be employed. Consequently, false positives and false negatives are generated through the applied multivariate analysis.
False negative results have the potential to miss important biomarker candidates with no recourse to find them later, while false positives lead to much wasted time, effort and resource investigating false biomarker candidates. Furthermore, it’s worth repeating that, at all times, the evidence of real biomarker candidates is actually there in your data and waiting to be found. At the outset, you carefully design the experiment, select subjects and prepare samples which you run on the best equipment available after painstakingly optimizing the running conditions… only to find your results underwhelming or, even worse, misleading!
Progenesis QI has a solution to ensure that missing values don’t jeopardize your chances of research success. Progenesis QI’s unique co-detection approach gives no missing values, and increases statistical power because you throw nothing away: we do a data extraction in which ion signals are perfectly overlaid before detection takes place by aligning the ions in retention time. This compensates for any retention time differences between samples. The overlaid ions can then be co-detected so that a single detection pattern is created for all the samples in the experiment, resulting in 100% matching of ions and no missing values!
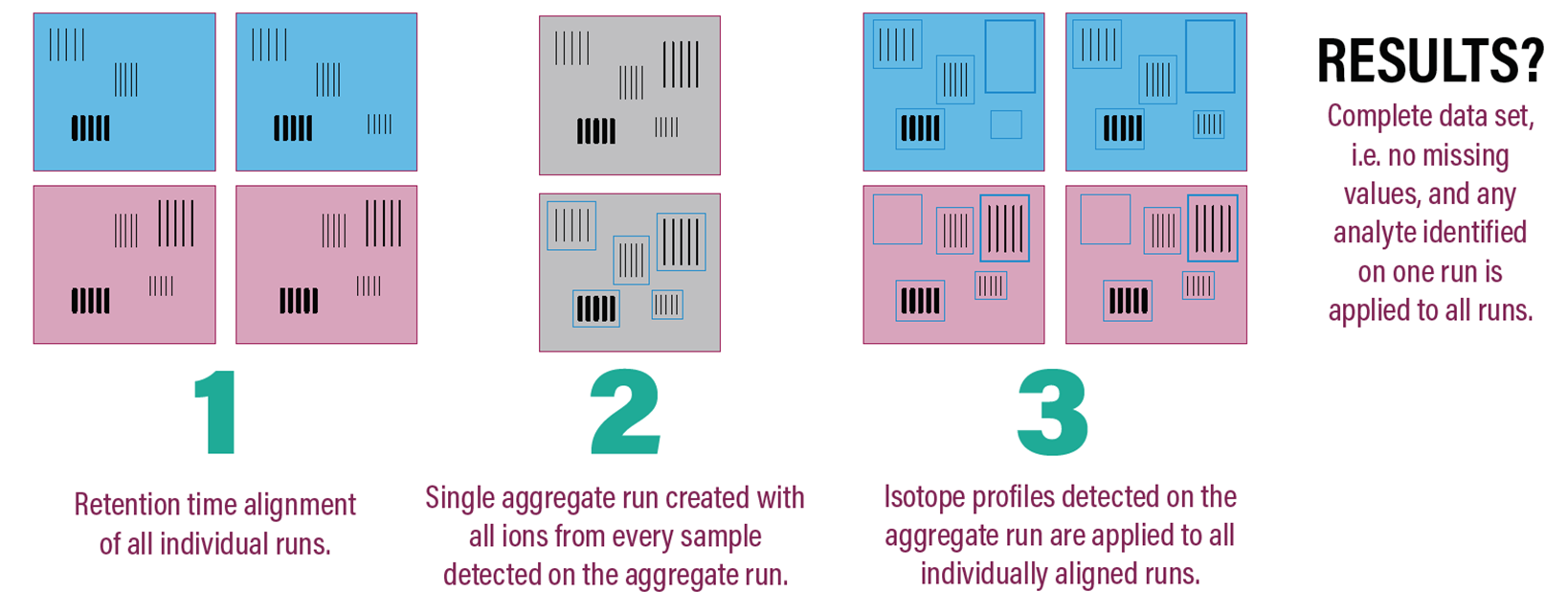
Consequently, as long as the differences are present in your data file, Progenesis QI will maximize your chances of picking them up. Working on data with no missing values improves the experimental specificity, sensitivity and therefore reproducibility of your research, allowing you to quickly and – more importantly – confidently quantify the compounds or proteins of interest.





